유전체 데이터 압축
무손실 유전체 데이터 압축
Enancio의 기술로 유전체 데이터 스토리지 및 전송 비용 절감

유전체 데이터 압축의 이점
Illumina는 혁신적인 시퀀싱 기술을 제공하고, 고객이 증가하는 차세대 시퀀싱(NGS) 데이터 출력을 관리할 수 있도록 지원하기 위해 최선을 다하고 있습니다. 이전에 Lena로 알려졌고 현재는 원본 리드 아카이브(ORA) 압축으로 알려져 있는 Enancio의 무손실 유전체 데이터 압축 기술은 최적의 속도와 효율성을 제공합니다.
유전체 데이터 압축은 다음을 가능하게 합니다.
- 데이터 스토리지 비용 절감
- 고속 데이터 파일 전송
- 내부 네트워크 트래픽 감소
유전체 데이터 무손실 압축 기술
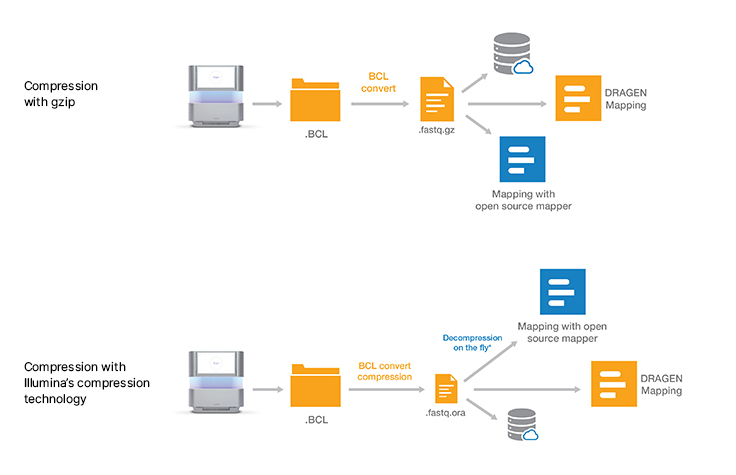
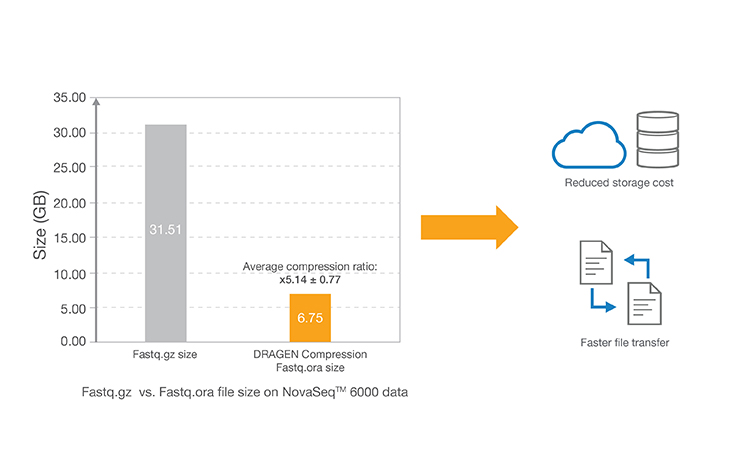
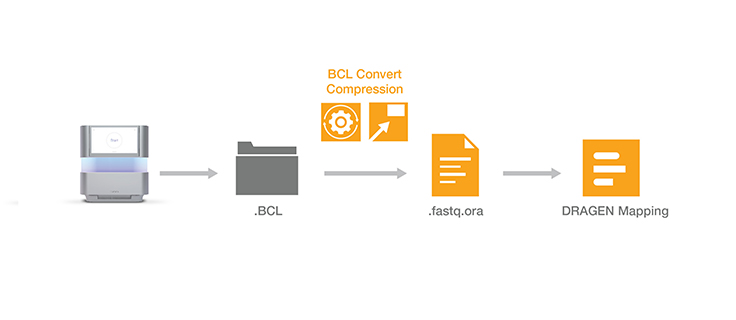
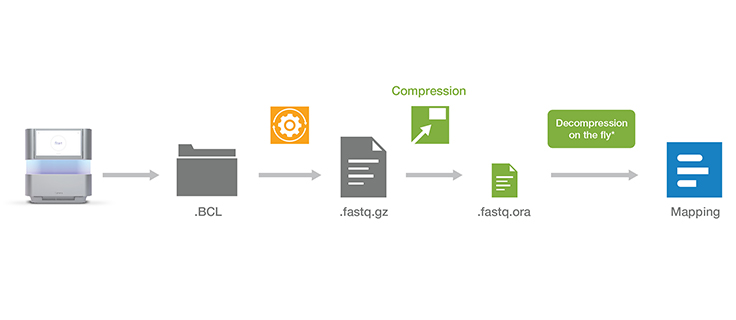
무손실 유전체 데이터 압축 기술은 Illumina 시퀀싱 시스템의 출력을 압축하여 데이터 스토리지 공간을 최대 5배까지 줄입니다. ORA 압축 기술은 참조 기반 압축 방법을 사용합니다. 아이디어는 초고속 매핑 기법을 사용하여 리드를 참조 유전체(reference genome)에 매핑한 다음, 해당 리드를 재생성하는 데 필요한 데이터(위치 및 차이점 목록)만 저장하는 것입니다.
다른 데이터 압축 기술은 일반적으로 속도가 느린 문제가 있습니다. ORA 압축 기술은 높은 압축 비율에 최적화되어 있으며, 신속한 압축 및 압축 해제와 동시에 데이터 무결성을 보존합니다. 품질 점수는 다양한 유형의 품질 체계에 맞게 조정된 범위 인코더 및 컨텍스트 모델을 사용하여 무손실 방식으로 인코딩됩니다.
DRAGEN ORA 압축 해제 소프트웨어 액세스
ORA 압축 기술로 압축된 모든 파일은 압축 해제 소프트웨어를 사용하여 쉽게 압축을 해제할 수 있습니다. 압축 해제 소프트웨어는 무료로 다운로드하여 사용할 수 있습니다.
압축 해제 소프트웨어 다운로드압축 해제 소프트웨어를 설치하면, 간단한 명령을 사용하여 즉시 압축 해제 출력을 BWA, STAR 및 Bowtie와 같이 널리 사용되는 광범위한 매핑 도구로 직접 보낼 수 있습니다. 압축 및 압축 해제 기술은 시퀀싱 데이터에 대한 정확하고 매우 빠른 분석을 제공하는 DRAGEN 2차 분석 소프트웨어에도 통합되어 있습니다.

기기 내 무손실 유전체 압축 가능
DRAGEN ORA 무손실 유전체 데이터 압축은 이제 NextSeq 1000 및 NextSeq 2000 시스템 및 NovaSeq X 시리즈를 사용하는 기기와 v3.8부터 시작하는 DRAGEN 2차 분석 서버에서 사용할 수 있습니다. 다음 사항에 대해 더 알아보세요.
NextSeq 1000/2000 System
NovaSeq X 시리즈
DRAGEN secondary analysis
압축 기술 FAQ
관련 솔루션
유전체 데이터 보관 및 보안
빠른 속도로 작동하도록 설계되었으며 규모에 따라 유연하게 조정이 가능한 클라우드로 대규모 유전체 및 NGS 데이터 세트를 안전하게 보관, 처리 및 공유하세요.
시퀀싱 데이터 분석
Illumina 시퀀싱 데이터 분석 소프트웨어는 귀하가 연구에 더 많은 시간을 사용하고, 분석 워크플로우를 구성하고 실행하는 시간을 줄이는 데 도움을 줍니다.
Illumina 인포매틱스 제품 포트폴리오
유전체 데이터 분석 및 관리를 간소화할 수 있는 광범위한 인포매틱스 제품을 살펴보세요.
압축 기술에 대한 질문이 있으십니까?
자세한 내용은 당사에 문의하십시오.
참고 문헌(References)
- NextSeq 1000 및 NextSeq 2000 시스템과 NovaSeq 6000 시스템에서 생성된 파일입니다.
- 이 결과는 NovaSeq 6000 시스템에서 30x 커버리지로 시퀀싱된 DNA 샘플 NA12878에서 얻은 것입니다. 데이터는 이 BaseSpace 프로젝트에서 접근할 수 있습니다: basespace.illumina.com/s/3ExEZMlH8Lkq.
- Li H. and Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009 Jul 15; 25(14): 1754–1760.
- Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan; 29(1): 15–21.
- Langmead B. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 2009 10:R25