Abstract
The choice of the human reference genome for mapping and variant calling has a direct impact on accuracy. This choice extends beyond a simple decision of GRCh37/hg19 or GRCh38/hg38. There are multiple versions of each reference, some include ALT and/or Decoy contigs, others don’t, some include portion of the genomes not covered in other versions. The final choice can impact the ability to call a variant. This can be illustrated by a recent update concerning the CBS, CRYAA and KCNE1 genes: it was noted that there is one copy of the gene in GRCh37/hg19 but there is a false duplication in GRCh38/hg38 which effectively leads to ambiguous mapping only in GRCh38/hg38. In 2021, Genome in a Bottle (GIAB) proposed using masking of the duplicate region to effectively remove false duplications for a set of concerned genes and thereby improve variant calling accuracy [1].
A separate and complementary approach to improving accuracy was used by the DRAGEN team in 2020. DRAGEN developed a graph-based genome to improve the mapping accuracy of Illumina reads in “Difficult-to-Map Regions” of the genome. The improvement was confirmed at the variant calling level and showed best winning accuracy in the ‘Difficult to Map Regions’ and “All Benchmark Regions” of the PrecisionFDA Truth Challenge v2 [2]. The graph genome includes several hundred thousand short, alternate contigs derived from phased population haplotypes added to the GRCh38/hg38 reference. This effectively creates yet another version of the GRCh38/hg38 human reference.
There is a lot of benefit to improve variant calling further by enhancing the reference while not impacting any downstream annotation. Especially when it comes to improving the reference in regions covering medically relevant genes. But we do recognize that every newly introduced flavour of the reference can create confusion for users because the term ‘GRCh38/hg38’ can no longer be used to fully describe the reference which is used in the analysis. In this article, we attempt to bring clarity on the various flavours of the GRCh38/hg38 reference, describe our current recommendation of which reference to use with DRAGEN as well as discuss additional planned improvements.
Recommended GRCh38/hg38 Reference Versions
High level, two versions of GRCh38/hg38 are currently recommended for use with DRAGEN 3.9, the hg38-alt-masked (non-graph) and hg38-alt-masked-graph. They are both available to download [3]. Both of these references contain the alt-masked functionality that was introduced in DRAGEN v3.9 and is described in more detail below. This new alt-masked functionality provides slight accuracy improvements over the previously recommended liftover-based ALT-aware references, the hg38-alt-aware (non-graph) and hg38-alt-aware-graph, shown below in Table 1.

Table 1: Recommended GRCh38/hg38 reference versions for use in DRAGEN, for non-graph and graph runs. Note that the baseline assembly for all these references was downloaded from [4].
The hg38-alt-masked reference is available to download [3]. The hash table can also be built using the following command.
dragen --ht-reference hg38.fa --ht-alt-aware-validate=true --ht-num-threads=40 --build-hash-table=true --ht-build-rna-hashtable=true --enable-cnv=true --ht-mask-bed /opt/edico/fasta_mask/hg38_alt_mask.bed --output-directory hg38_alt_masked
The alt-masked hash table creation is supported from DRAGEN 3.9 onwards but a 3.9 alt-masked hash table can be used with older versions of DRAGEN.
The hg38-alt-masked-graph genome hash table is available to download [3]. The hg38-alt-masked-graph hash table is compatible with pre-3.9 versions of DRAGEN. DRAGEN does not support the users building their own custom graph genomes. This is because altering the population haplotypes can cause an accuracy regression if the new haplotypes compete with other regions of the genome. Great care needs to be taken in building a graph genome and as such a fully automated approach is not currently supported in DRAGEN.
Note regarding compatibility between previously built BAMs and new Variant Calling runs: It is important to keep the same reference between the mapping/alignment stage and the variant calling stage. Hence if a BAM was mapped with “*alt-aware” then the Variant Calling should also use alt-aware and same applied to alt-masked. If a user wants to update their results with alt-masked, it is suggested to remap the BAM with alt-masked prior to variant calling with alt-masked.
Common Reference Assembly as the baseline to all GRCh38/hg38 versions
All versions of GRCh38/hg38 reference recommended for use in DRAGEN (cf. Table 1) use a common FASTA assembly as their baseline, downloaded from [4]. Then, the versions are differentiated by whether the FASTA assembly is augmented by a set of population SNP and/or set of population-derived haplotypes of phased variants, offering alternate paths to the linear reference. This yields the graph-based versions, with more details given in [2]. In addition, the native GRCh38/hg38 ALT contigs, included in the baseline assembly, can be handled either by the “alt awareness” capability of the DRAGEN mapper (in alt-aware versions) or the new alt-masked functionality (in alt-masked versions).
The command FASTA assembly contains 3366 different contigs as described in the table 2 below.

Table 2: Components of the common FASTA assembly, downloaded from [4].
Handling of native GRCh38/hg38 ALT contigs: ALT-aware versus ALT-masking
Prior to the 3.9 release, DRAGEN employed an ‘ALT-aware' alignment liftover procedure to leverage the alternative (ALT) contigs present in some hg19 and hg38 FASTA files. This method uses a liftover SAM file containing alignments of ALT’s to the primary chromosome contigs. These alignments were produced by the Genome Reference Consortium. Reads aligning best to an ALT contig region with a valid liftover are lifted to the corresponding chromosomal location. Reads aligning best to an ALT region with no valid liftover remain mapped to the ALT contig.
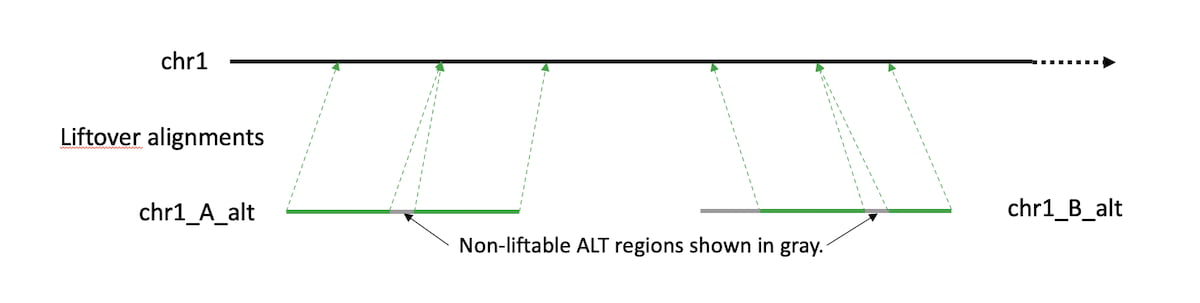
Figure 1: Example of a correct liftover between alt region and main reference.
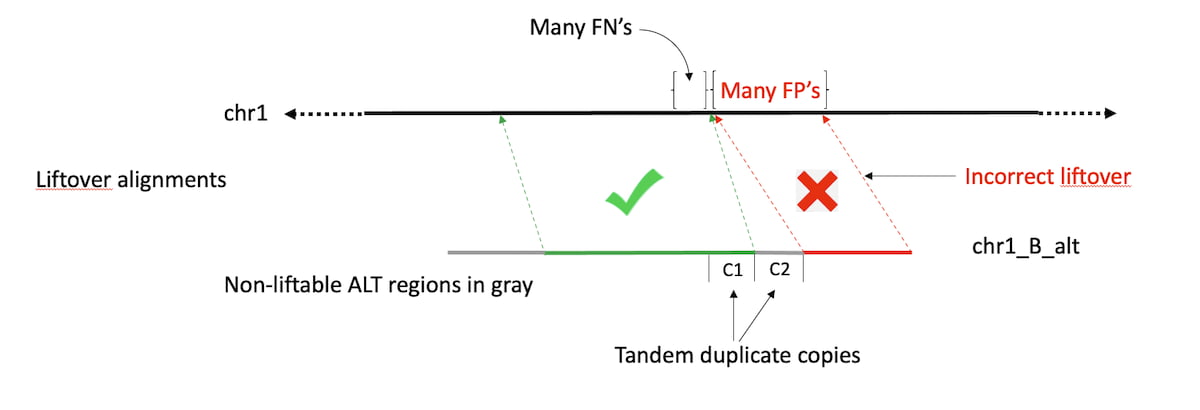
Figure 2: Incorrect Liftover alignments can create extra FPs.
DRAGEN has adopted a new approach to handle native reference ALT contigs, where strategic positions of the ALT contigs are masked to increase accuracy. Segments which are similar to the primary assembly are masked, so they do not compete and steal alignments or squash MAPQs. Segments which are quite different are left unmasked, functioning essentially as decoy sequences. The masking status of several marginal regions is assigned based on empirical impact on mapping accuracy and variant calling. As demonstrated in Figure 3 in the results section, DRAGEN’s ALT masked references deliver improved variant calling accuracy vs. the liftover-based ALT-aware method. The base masking approach has the benefits of using ALT contigs without the negative consequences. It is also easier to define, maintain and improve. The masks will likely continue to get refined over time, but they've already surpassed liftover-based performance.
DRAGMAP, which is the open source version of the DRAGEN mapper released in collaboration with Broad (https://github.com/Illumina/DRAGMAP) does not support the liftover-based ALT awareness [5]. Instead, DRAGMAP uses the same recommended alt-masked approach that is used in the latest version of DRAGEN. This means that it also has the accuracy improvement of the masking approach compared to the older liftover approach and liftover support is not needed in DRAGMAP.
Graph Based Reference
As mentioned earlier, DRAGEN also supports a graph-based reference to improve the mapping accuracy of Illumina reads in the difficult-to-map regions of the genome. The graph functionality does not use native GRCh38/hg38 ALT contigs but uses carefully chosen population haplotype segments which usefully distinguish among homologous regions, and provide alternate paths known to the population to the linear reference.
Mapping difficulty can arise because a region (such as MHC) is highly polymorphic, and sample reads differ so greatly from the reference genome that a mapper can’t find or recognize a good match. Much more commonly, though, mapping difficulty arises when sample reads match a region reasonably well – but also match other regions almost or just as well. This happens when near copies of a region appear in several places in the reference genome (segmental duplicates), or in the case of common highly repetitive sequences.
In many cases, such mapping difficulty can be overcome by utilizing known variation patterns in the population, rather than just a single reference genome. Suppose a short read (or read pair) matches two regions, A and B, equally well but imperfectly, with two nucleotide differences from the reference genome in each region. Based on the reference alone, a mapper can only pick A or B at random and align there with zero MAPQ. But suppose we know that the read’s two differences from region A occur commonly in the population, while its two differences from B have not been observed in the population. We can use this knowledge as a guide to map the read to region A, with reasonably high confidence.
The choice of population haplotype segments is key to improving the mapping in the difficult parts of the genome. Increasing the number and population diversity can improve the accuracy further but this can have a negative impact if the haplotypes end up competing with each other and result in ambiguous read mappings. Mask-based hg38 ALT-awareness also plays better with graph references, keeping out of the way to allow graph-path liftover to guide mapping without interference.
Graph references are currently only supported on hardware accelerated versions of DRAGEN and therefore not supported with DRAGMAP.
Measuring the performance of the references
The impact of the alt-masked approach versus the liftover-based alt-awareness is shown in Figure 3 below, for both the non-graph and graph reference versions, using the NIST v.4.2.1 truth set [6]. The alt-masking approach leads to a reduction in FP+FN numbers for both snps and indels and in both the graph and non-graph genomes.
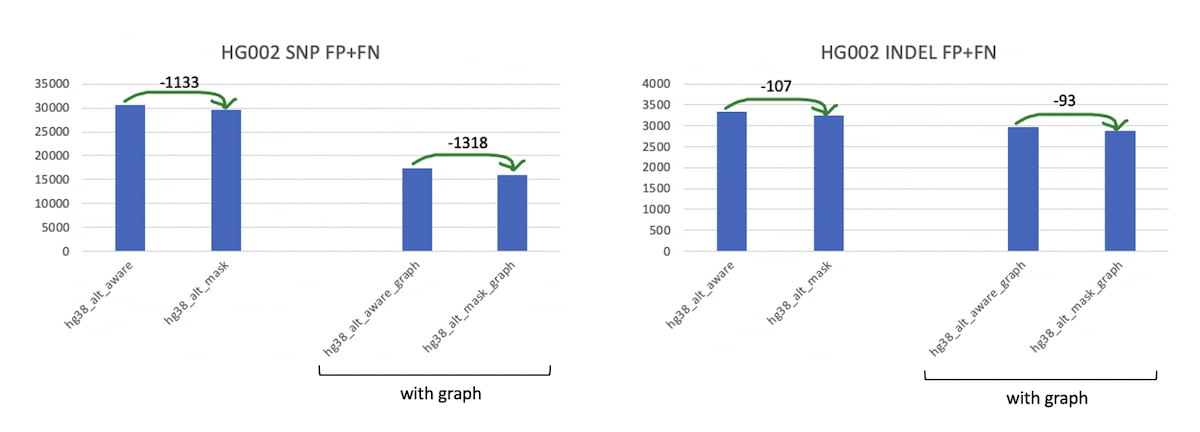
Figure 3: Comparison of alt-masked vs alt-aware accuracy, using NIST 4.2.1 truth set.
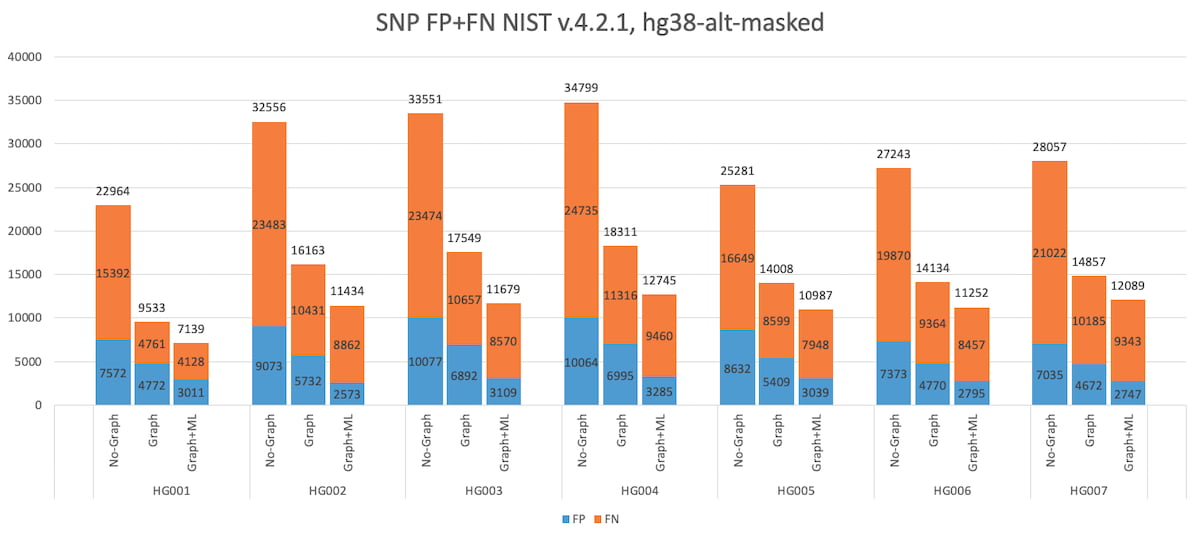
Figure 4: HG001 through HG007 SNP Accuracy Results in the extended truth set (v4.2.1 VCF and BED).
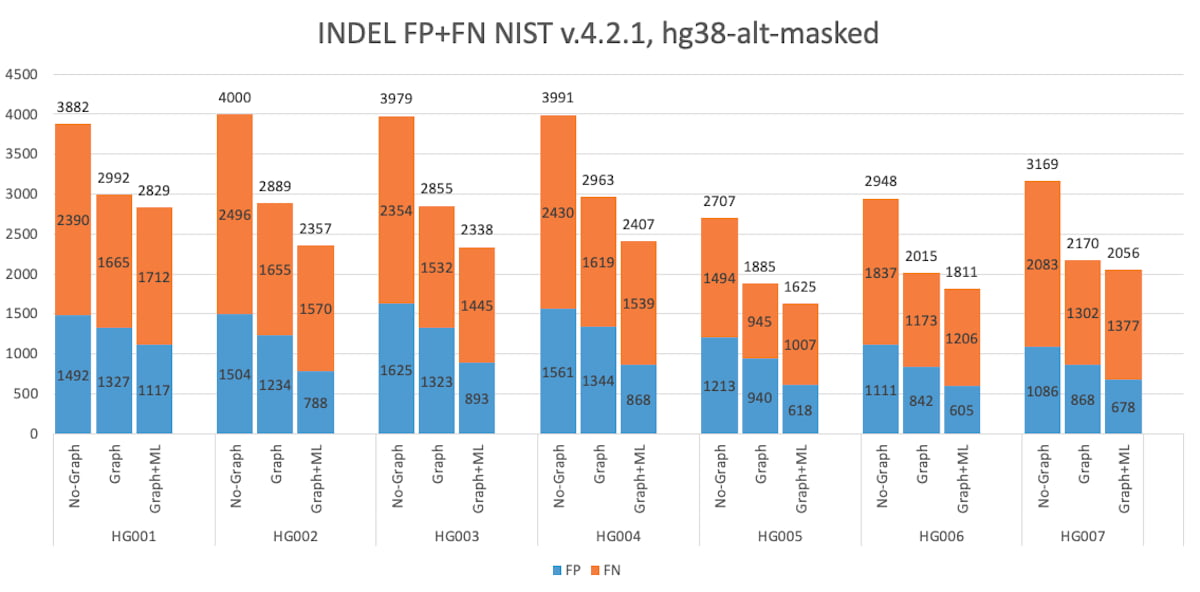
Figure 5: HG001 through HG007 INDEL Accuracy Results in the extended truth set (v4.2.1 VCF and BED).
It is important to use the latest and most comprehensive truthsets when measuring the impact of graph reference (or other reference improvements). For example, the 50% reduction in total errors from DRAGEN graph is measurable with the extended truth set v4.2. This performance gain is not apparent when benchmarking against the older v3.3.2 truth set. That is partly because the v3.3.2 truth set does not include the difficult to map regions, but also because the v4.2 truth set corrects errors that were present in v3.3.2. Indeed, DRAGEN graph appears to yield additional SNP and INDEL false positives (FP) compared to legacy DRAGEN when using the older truth set v3.3.2. However, a large proportion of the additional FP calls are due to the v3.3.2 truth VCF being incomplete. The same ‘FP’ variants are marked as true positives in the v4.2 truth VCF.
Further GRCh38/hg38 Reference Improvements (adopted by DRAGEN beyond 3.9 release)
The full potential of both genome masking and graphs has not yet been reached in terms of accuracy. The Genome in a Bottle (GIAB) Consortium, Genome Reference Consortium (GRC) and Telomere-to-Telomere (T2T) Consortium have also contributed to improvements of the GRCh38/hg38 reference genome [1], with two types of improvements: 1) masked bases in the primary assembly to remove false duplication; 2) include new Decoy contigs. The DRAGEN team is currently in the process of evaluating these changes, and incorporating them into the most recent reference versions. Table 3 shows a roadmap on how we plan to release the reference updates in future DRAGEN releases. The newly masked bases will be incorporated first, and updated references will be name *alt-masked-V2*. The new Decoys are not fully finalized just yet and will be incorporated in a further release.

Table 3: Recommended GRCh38/hg38 reference versions for use in DRAGEN, for non-graph and graph runs. Note that the baseline assembly for all these references was downloaded from [4].
More details on the newly masked bases: towards alt-masked-V2
The GIAB consortium has recently released a new reference which masks false duplications in GRCh38/hg38 [1]. GIAB worked together with the GRC to develop a list of regions in GRCh38 that could be masked without changing coordinates or harming variant calling, because they were erroneously duplicated sequences or contaminations. These duplicated regions were identified by the T2T [7]. The newly masked bases include portions of chr21 which resulted in mapping improvements in some key medical genes CBS, CRYAA and KCNE1.
Figure 6 below is taken out of the Supplementary Material of [1], and shows how the variant calls across technologies are consistent for the GRCh37 reference, because GRCh37 does not contain the false duplication. GRCh38 contains a false duplication of part of the gene, so many reads mismap to the false copy of the gene, KCNE1B.

Figure 6: KCNE coverage across different technologies with GRCh37 and GRCh38 (without the newly masked bases) [1].
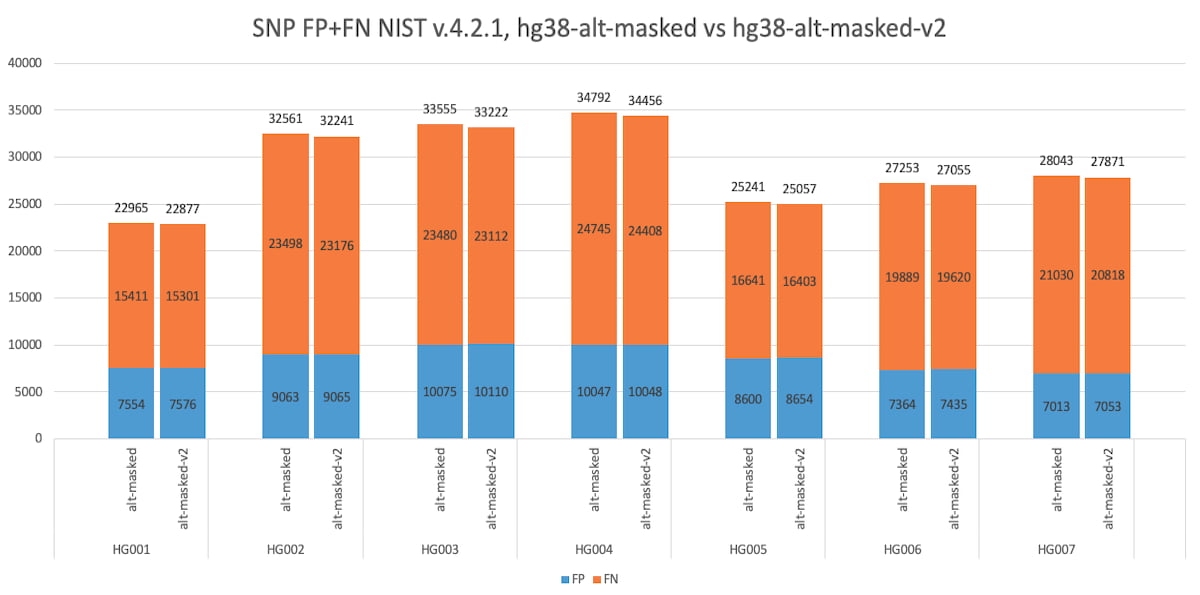
Figure 7: Accuracy Improvement using the GIAB masked reference, assessed over the whole genome, using V.4.2.1 NIST truth set. hg38-alt-masked, versus hg38-alt-masked-V2, for SNPs.
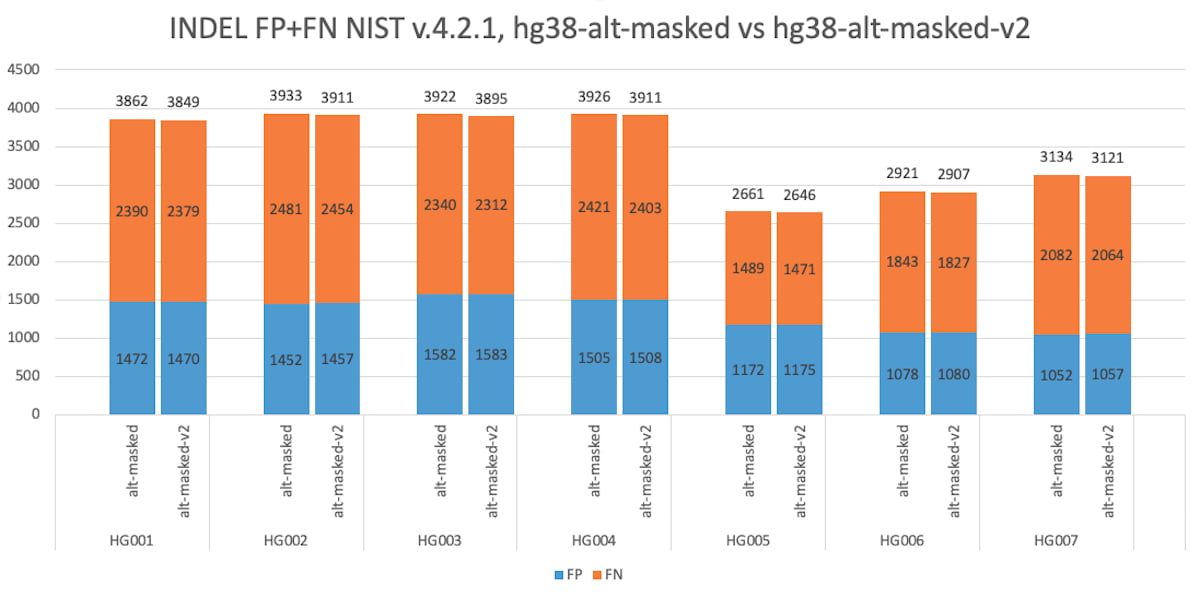
Figure 8: Accuracy Improvement using the GIAB masked reference, assessed over the whole genome, using V.4.2.1 NIST truth set. hg38-alt-masked, versus hg38-alt-masked-V2, for indels.
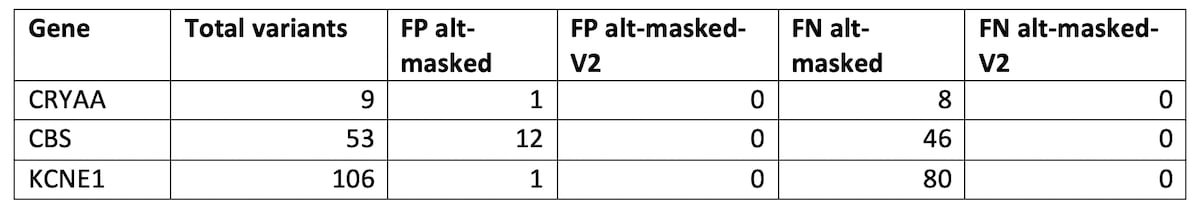
Table 4: HG002 SNP accuracy improvement on genes affected by the GIAB masking, using the CMRG v.1.0.0 truth set, comparing hg38-alt-masked and hg38-alt-masked-V2.
More details on the upcoming new Decoy contigs: towards alt-masked-V3
A second approach which can improve the accuracy of the mapping and variant calling is through the use of decoy sequences. A joint effort between the GIAB and Baylor College of Medicine [8] is currently assessing augmenting the GRCh38/hg38 reference with new decoys to correct for regions which have been falsely collapsed in the reference e.g., the reference contains a single copy of a region whereas it should contain a second homologous region. The decoys, which are similar but not identical, will remove false positive variants by providing alternative mapping locations instead of forcing the reads to map to the wrong copy of the sequence.
The third planned improvement to the references is the introduction of new graph alt contigs based on an expanded set of population haplotypes. The current graph genome used by DRAGEN has already demonstrated great capability to improve accuracy. There are opportunities to further extend the list of population-based haplotypes to cover a more diverse set of genome regions and/or ethnicities. The goal is to build a single graph genome which is capable of maximising accuracy across all populations. This will remove the need to either use a graph which is not optimal for an individual or generate a graph reference which is population specific.
Creating A Custom Masked Reference
The hg38-alt-masked reference is available to download [3]. The hash table can also be built using the following command.
dragen --ht-reference hg38.fa --ht-alt-aware-validate=true --ht-num-threads=40 --build-hash-table=true --ht-build-rna-hashtable=true --enable-cnv=true --ht-mask-bed /opt/edico/fasta_mask/hg38_alt_mask.bed --output-directory hg38_alt_masked
Please note that starting from DRAGEN 3.9, if no liftover or masked bed is specified on the HT building command line, the default behavior of DRAGEN is to automatically apply the alt-masked bed, to generate hg38-alt-masked (or hg19-alt-masked) reference by default. The alt-masking does not apply to GRCH37 or hs37d5, since those references do not include ALT contigs natively.
DRAGEN provides the functionality to make custom masked genomes by modifying the hg38_alt_mask.bed file packaged with DRAGEN or even by the user creating one. DRAGEN will create a hash table by treating any position contained in the bed file as an N in the FASTQ for mapping purposes. This essentially creates the same hash table as modifying the reference directly. If there are contigs in the bed file which are not in the FASTA then DRAGEN 3.9 will abort. In future versions, DRAGEN will not abort and only mask the regions which are present.
Note on hg19:
hg19 also contains a set of native ALT contigs, so the discussion of alt-aware vs alt-masked apply to hg19 in the same way as they were described above for hg38.
Summary of Results and Acknowledgement
As the assessment of mapping and small variant calling accuracy of various reads technologies extends into more complex regions of the genome, it is critical to keep maintaining and updating the reference genome, with new and improved versions to keep up with the discoveries and areas of improvements. We have described here the various improvements of the reference genome that have already been released as part of past DRAGEN releases (3.9 and earlier), as well as shed some light on further upcoming improvements, which will be included in future DRAGEN releases.
We would like to thank the efforts of the Genome in a Bottle (GIAB) Consortium, Genome Reference Consortium (GRC) and Telomere-to-Telomere (T2T) Consortium for their continued contributions to improving the reference genome, which in turn become mapping and variant calling accuracy improvements. This is especially important in challenging medically relevant genes, as it helps variant discovery and supports medical decision to improve human health.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
References
- Wagner et al. Towards a Comprehensive Variation Benchmark for Challenging Medically-Relevant Autosomal Genes. BioRxiv (2021) doi:2021:09.07
- DRAGEN Wins at PrecisionFDA Truth Challenge V2 Showcase Accuracy Gains from Alt-aware Mapping and Graph Reference Genomes
- https://support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform/product_files.html
- http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa
- https://github.com/Illumina/DRAGMAP/
- Wagner et al. Benchmarking challenging small variants with linked and long reads. BioRxiv (2021) doi:2020:07.24
- Nurk et al. A complete reference genome improves analysis of human genetic variation. BioRxiv (2021) doi: 2021.05.26.445798
- Publication pending.