Introduction
Structural variants (SVs) contribute to a significant proportion of nucleotide sequence differences between individuals and are implicated in many human diseases1, 2. Yet, the accurate detection of SVs remains a challenge partly because significant deviations from the reference genome can make alignment difficult especially for short-read whole-genome sequencing (WGS) data3. Although recent advances in long-read sequencing technologies have made it easier to detect SVs, their application is limited by cost, throughput and requires a significant amount of DNA (~5ug per genome, versus ~25ng in Illumina PCR-free protocol)4. Alternatively, scientists can study SVs at the population level by genotyping known variants using graph methods built from a reference SV database that could be built using a combination of technologies5. As these reference databases evolve, the population sequencing data can be re-genotyped with the updated information for a more complete picture of SV variation.
In this article, we assess the genotyping performance of our graph-based genotyper, Paragraph, that is capable of genotyping known SVs in a large population of samples sequenced with short reads. Paragraph constructs a directed acyclic graph with paths representing the reference allele and any possible alternative alleles, realigns reads to this sequence graph and genotypes the SVs based on the realigned reads (Figure 1). We applied Paragraph to a breakpoint-accurate SV truth set that was built from three samples sequenced with long reads6. In addition to assessing the accuracy by genotyping the SVs in the samples used to build the truth data, we genotyped our truth set SVs in the 1000 Genome population of 2,501 unrelated individuals and used population genetics metrics to quantify the accuracy of the SV calls7,15. Metrics such as Hardy-Weinberg equilibrium (HWE) allows us to study the overall performance of a variant in the population and to identify possible systematic errors that might not be detected by just testing a handful of samples8. Using the 1000 Genome samples, we demonstrate that the population-level genotyping statistics can help to clean the calls. We further quantify the demographic distribution of these SVs in different ethnicities and identified signatures of purifying selection in functional genomic elements.
Results
Building the SV truth set
We start from three samples that are included in the Genome in a Bottle (GIAB) project data: NA12878 (HG001), NA24385 (HG002) and NA24631 (HG005) (Availability of Data and Materials). Long-read sequence data was generated for these samples using a Pacific Bioscience (PacBio) Sequel system. These samples were sequenced to an average of 30-fold depth with ~11,000 base pair (bp) HiFi reads. From this data, we called SVs (50bp+) using PBSV9. After merging these SVs, we identified 38,709 unique autosomal SVs. Because Paragraph is designed to work in regions with a single SV, we excluded the regions where more than one SV was observed (eg, overlapping or nearby SVs with different break points). This left 20,108 SVs (9,238 deletions and 10.870 insertions) as our long-read ground truth (LRGT) for testing and for population genotyping purposes. A complete description of these SVs can be found here6.
Single-sample recall and precision
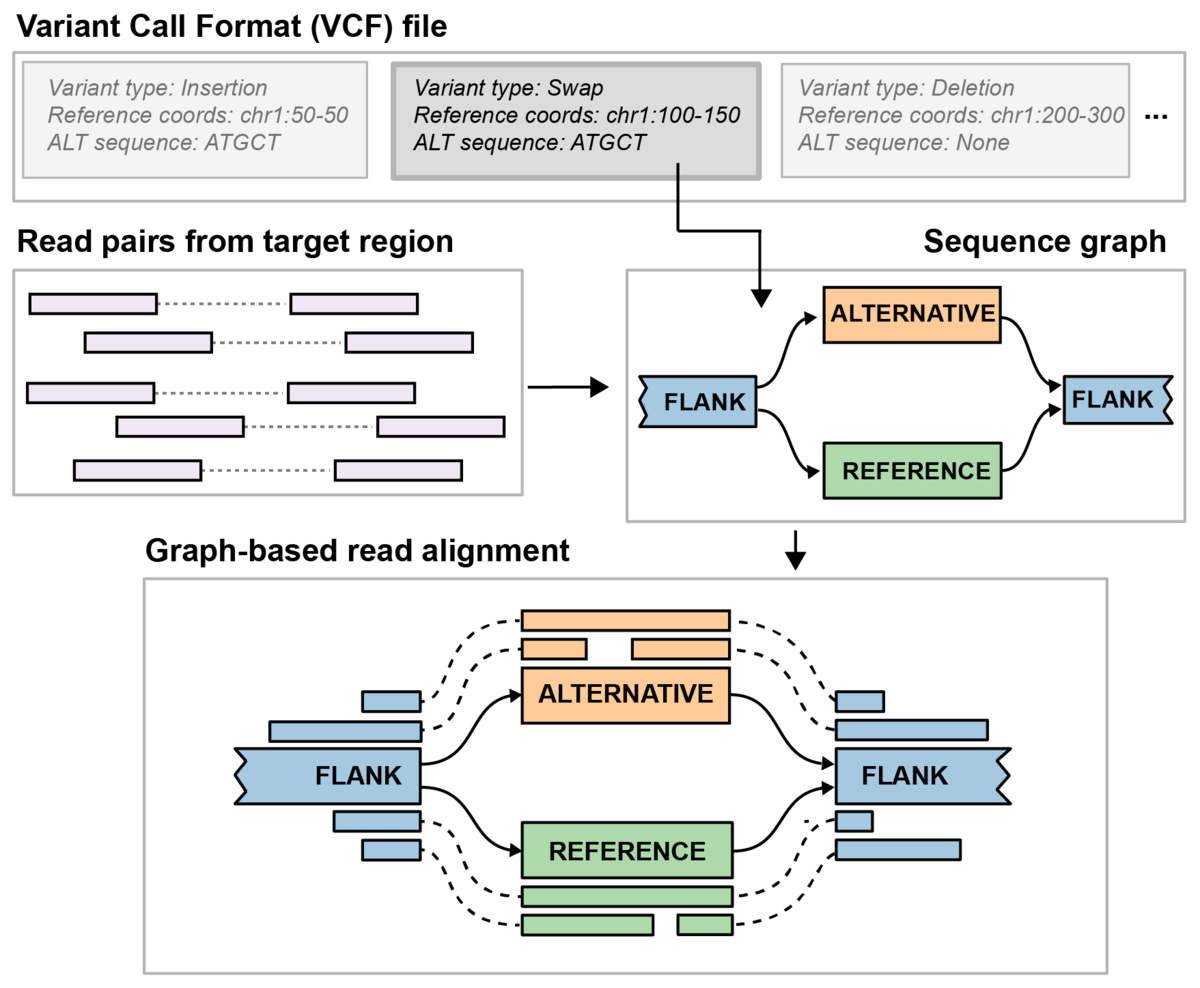
To evaluate the genotyping performance of these SVs, we genotyped our LRGT SVs on short-read data of NA12878 (63x), NA24385 (35x) and NA24631 (40x) using our graph genotyper, Paragraph (Figure 1)6. Because the accuracy of classifying homozygous versus heterozygous alleles in the long-read SV caller has not been systematically evaluated yet, we focus our test on the presence/absence of variants and not genotyping concordance. Thus, we define a variant as a true positive (TP) if Paragraph makes a non-reference genotype call where an SV is also present in the LRGT and a false positive (FP) if Paragraph calls a non-reference allele at a location where LRGT does not include a variant in that sample. With this definition, the LRGT has 38,239 individual alternative genotypes that serve as TPs and 22,085 reference genotypes to calculate FPs. Averaged on the independent tests of three samples, Paragraph has a recall of 0.84 and a precision of 0.88 (Table 1)6.
Type |
#Truth TPs |
Recall |
#Truth FPs |
Precision |
F-score |
Deletion |
16,936 |
0.84 |
10,778 |
0.92 |
0.88 |
Insertion |
21,303 |
0.88 |
11,307 |
0.89 |
0.88 |
Table 1. The performance of Paragraph, tested on LRGT
Looking into the problematic calls, we found that 59% of the FNs and 77% of the FPs occur in SVs that overlap within tandem repeats (TRs). Paragraph has a much better recall (0.90) in SVs that are outside of TRs than in those that are within TRs (0.79). We also found that smaller (<200bp) SVs are much more likely to be within TRs (~75%) than larger (>1,000bp) SVs (~35%). In addition, in our original paper, we show that errors in SV breakpoints (e.g. incorrect location or size) will negatively impact the performance of Paragraph.
Genotyping 2,501 diverse human genomes
Genotype results from many unrelated samples in a population allows us to assess the accuracy of a variant caller with population genetics measurements such as HWE, even when no ground truth data exists. As a demonstration for Paragraph genotyping capability, and also to study population statistics, we genotyped our LRGT SVs in 2,501 unrelated individuals (not including NA24385 or NA24631) from the publicly available 1000 Genome sequencing resource. This dataset represents an ethnically diverse population with 660 Africans (AFR), 347 Americans (AMR), 501 East Asians (EAS), 489 South Asians (SAS) and 504 Europeans (EUR)10. All samples were sequenced on Illumina NovaSeq platforms with 150 bp paired-end reads to at least 30-fold depth. The sequencing data was processed with our DRAGEN v3.5.7b workflow14.
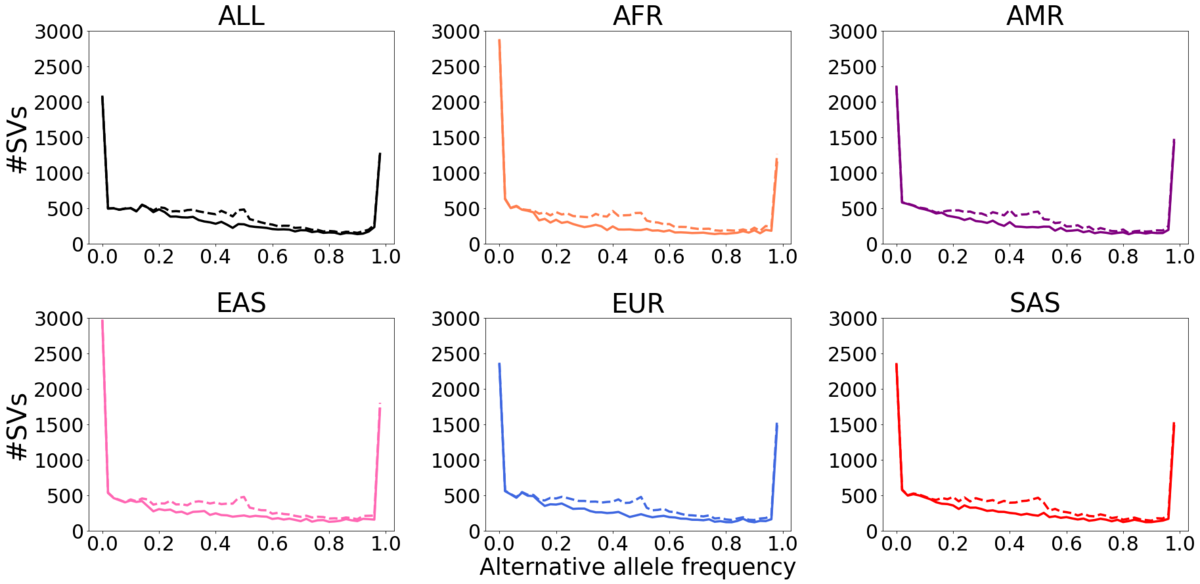
Dashed lines: all SVs. Solid lines: SVs that pass HWE test in at least one population.
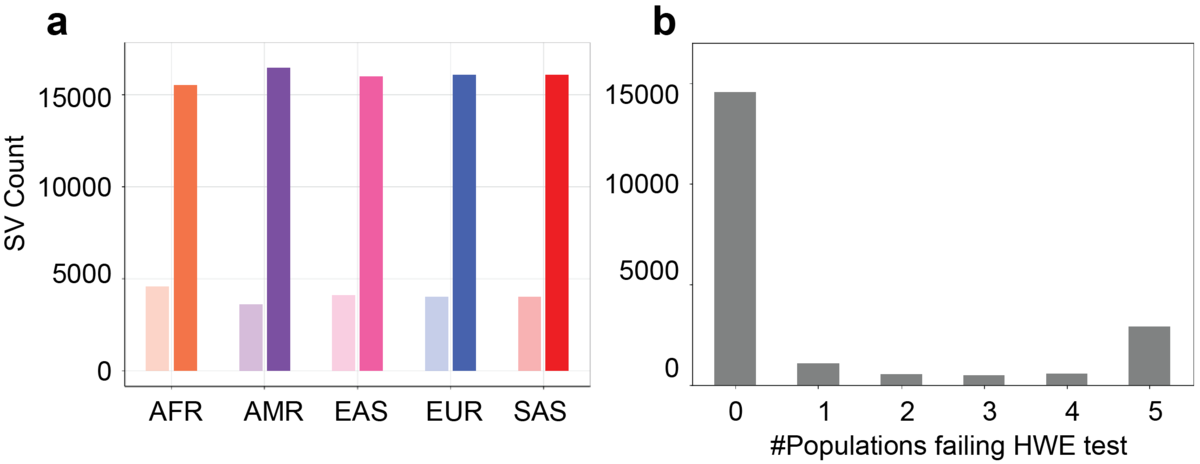
a) Count of HWE passed (solid bars) and failed (light bars) SVs in each population. b) Count of how many populations failed the HWE test for the SVs that failed the HWE test in at least one population.
We observed that 14,568 (73%) of the SVs pass the HWE test in all five populations. 2,904 (14%) of the SVs do not pass the HWE test in any population. 2,636 (13%) of the SVs pass the HWE test in some populations but not all five (Figure 3b). Of the SV that fail the HWE test in all five populations, 78% overlap TRs. In TRs, the different paths through the genome graph can be very similar and that makes the current alignment employed in Paragraph nearly stochastic, leading to a high number of heterozygous genotypes. This suggests that a different graph genotyping model is needed to improve the genotyping performance of TR-overlapping SVs. After removing the HWE-inconsistent SVs, we note that the unexpected AF peak at around 0.5 is eliminated (Figure 2, solid lines). For the 14% SVs that were HWE-inconsistent in some of the populations, most do not show extremely significant HWE p-values, possibly indicating these SVs could have breakpoint deviations in a few samples or in some populations.
SV distributions in the mixed population
After filtering out the SVs that failed our HWE test, the overall AF distribution is similar across all populations. Despite this similarity in AF, the principal component analysis (PCA) on genotype dosages of HWE-passing SVs (0 for homozygous reference genotypes and missing genotypes, 1 for heterozygotes and 2 for homozygous alternative genotypes) reveals population-specificity of the LRGT SVs (Figure 4a). Projected in the two-dimensional space of the first and second principal components, the AFR, EAS, EUR and SAS samples are clearly separated. Conversely, the AMR samples map between the different ancestral populations as would be expected for an admixed population.
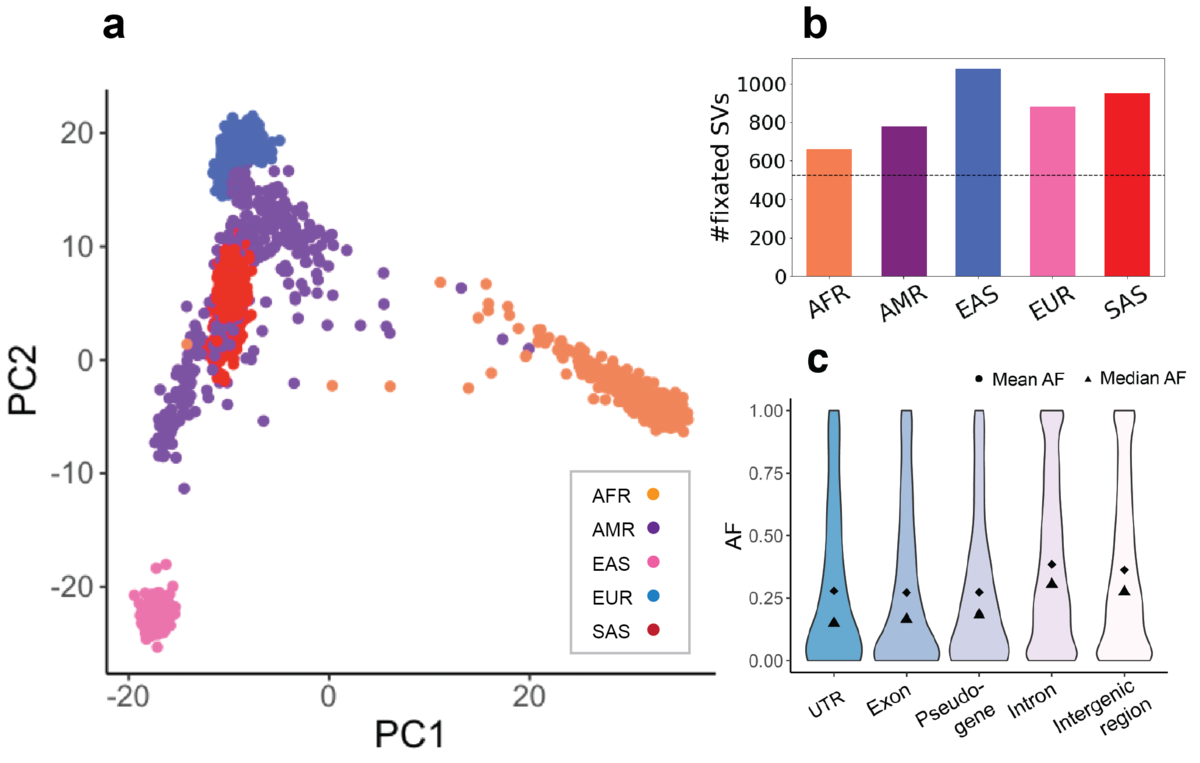
a) PCA biplot of 1000 Genome samples with HWE-passing SVs. b) Count of fixed SVs in each population. c) SV AFs in different functional elements across all populations.
In total we observed 525 SVs that are fixed in all 5 populations possibly representing rare alleles in the reference genome (Figure 4b). Besides these, there are 715 SVs that are fixed in at least one population. Consistent with being the most diverged population, AFR has fewer fixed SVs than the other populations. Combined with functional annotations, 303 fixed SVs are exonic. The 1,638 bp exonic insertion in UBE2QL1 was also reported at a very high frequency in two previous studies12, 13. Particularly, a recent study by TOPMed reported this insertion in all 53,581 sequenced individuals from mixed ancestries13.
We also observed different AFs for the SVs based on genomic context. SVs within exons, pseudogenes and untranslated regions (UTRs) of coding sequences, in general, have lower AFs than those in intronic and intergenic regions. SVs in introns and intergenic regions have more uniform AF distributions compared to the more extreme AFs in functional elements (UTRs, exons) (Figure 4c). All these suggest a purifying selection against SVs with potentially functional consequences7. Although common SVs are more depleted in functional elements, we do see a few common SVs within exons of genes including TP73 (AF=0.16 overall, tumor suppressor gene), FAM110D (AF=0.65 overall, functions to be clarified, possibly related with cell cycle) and OVGP1 (AF=0.20 overall, related to fertilization and early embryo development). Specifically, the deletion in TP73 has very different frequencies in the sub-populations: in AFR it is as low as 0.02 while in EUR and EAS it is higher than 0.20.
Conclusions
In this article, we briefly reviewed an initial version of an SV truth set that we created for three samples that are included in the GIAB consortium. This truth set demonstrates the ability to use graph methods to genotype a database of high-quality SVs. Our ongoing work with extend this database and improve Paragraph with better genotyping accuracy. By genotyping these SVs in the diverse 1000 Genome population, we demonstrated how population data can be used as a quality control even in the absence of single-sample ground truth data. Additionally, we demonstrated that many of the exonic SVs identified in our ground truth data are also found in the 1000 Genome samples and exhibit signs of selection. Genotyping these exonic SVs directly using Paragraph in cohorts with WGS may further help us understand the biology of these SVs/genes.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
Availability of data and materials
Paragraph software is publicly available at https://github.com/Illumina/paragraph.
NA12878, NA24385 and NA24631 PacBio data were deposited in SRA under PRJNA540705, PRJNA529679 and PRJNA540706. Their Illumina data was deposited in ENA under PRJEB35491.
The Dragen reprocessed 1000 Genome data is available at https://registry.opendata.aws/ilmn-dragen-1kgp/.
References
- Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J.O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet 14,125-138 (2013).
- Lee, C. & Scherer, S.W. The clinical context of copy number variation in the human genome. Expert Rev Mol Med 12, e8 (2010).
- Goodwin, S., McPherson, J.D. & McCombie, W.R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333-351 (2016).
- Ashley, E.A. Towards precision medicine. Nat Rev Genet 17, 507-522 (2016).
- Logsdon, G.A., Vollger, M.R. & Eichler, E.E. Long-read human genome sequencing and its applications. Genome Biol 20, 291 (2019).
- Chen, S. et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol 20, 291 (2019).
- Sudmant, P.H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75-81 (2015).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993 (2011).
- Wenger, A.M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37, 1155-1162 (2019).
- Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68-74 (2015).
- Wigginton, J.E., Cutler, D.J. & Abecasis, G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 76, 887-893 (2005).
- Sherman, R.M. et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet 51, 30-35 (2019).
- Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv, 563866 (2019).
- Chu E, Friedman A. DRAGEN reanalysis of the 1000 Genomes Dataset now available on the Registry of Open Data. Amazon Web Services, 2020.
- Bryska-Bishop, M. et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv, 430068 (2021).