Introduction
Alpha thalassemia and spinal muscular atrophy (SMA) are among the most prevalent carrier conditions worldwide. Deleterious copy number variants (CNVs) in their respective causative genes, HBA1/2 and SMN1, are responsible for around 95% of these carrier conditions. While next-generation sequencing (NGS) panel data has demonstrated good results and has broad adoption for CNV detection, both HBA and SMN genes occur in non-unique regions of the genome that are generally difficult to profile with standard sequencing and bioinformatics workflows due to high homology with a pseudogene or paralog. Due to such challenges, targeted PCR-based tests (for example, MLPA or qPCR) specifically designed to profile HBA and SMN loci are often used to supplement the NGS panel-based testing generally used in carrier screening research, adding significant additional expense and workflow complexity.
Targeted variant callers that accurately genotype HBA and SMN from Illumina whole-genome sequencing (WGS) data were released in DRAGEN v4.2 and v3.9, respectively. However, analysis of SMN and HBA from exomes was not feasible until now because standard exome panels did not cover the genomic regions leveraged in such callers, and because the depth normalization approaches initially leveraged in the callers was insufficient to handle capture efficiency biases inherent to enrichment-based assays.
Here, we present updated versions of SMN and HBA targeted callers, released in DRAGEN v4.4, as well as a spike-in panel (Illumina CS/PGx Custom Enrichment Research Panel) that can be used to supplement the Illumina DNA Prep with Exome 2.5 Enrichment panel to allow for accurate HBA and SMN genotyping on Illumina exomes.
Illumina CS/PGx Custom Enrichment Research Panel
Many DRAGEN targeted callers depend on coverage of non-exonic regions. In particular, some differentiating sites leveraged for SMN1/2 copy number calling and nonhomologous regions used in HBA copy number genotyping are non-exonic. The Illumina CS/PGx Custom Enrichment Research Panel was thus developed to ensure coverage of such regions for various challenging medically relevant genes. Genes with extended coverage in the spike-in panel are listed in Table 1. While the bioinformatics improvements needed to make use of the additional content have currently been released only for HBA and SMN, the addition of the Illumina CS/PGx Custom Enrichment Research Panel to the exome panel should enable users to take advantage of bioinformatics improvements planned in future DRAGEN releases for other genes in the panel.
The spike-in panel covers 359 kb of genome in total, which represents less than 1% of the genomic content covered in the Illumina DNA Prep with Exome 2.5 Enrichment panel. Therefore, when added to the exome panel, the spike-in panel has minimal impact on the overall level of coverage of exome targets with a fixed level of sample multiplexing.
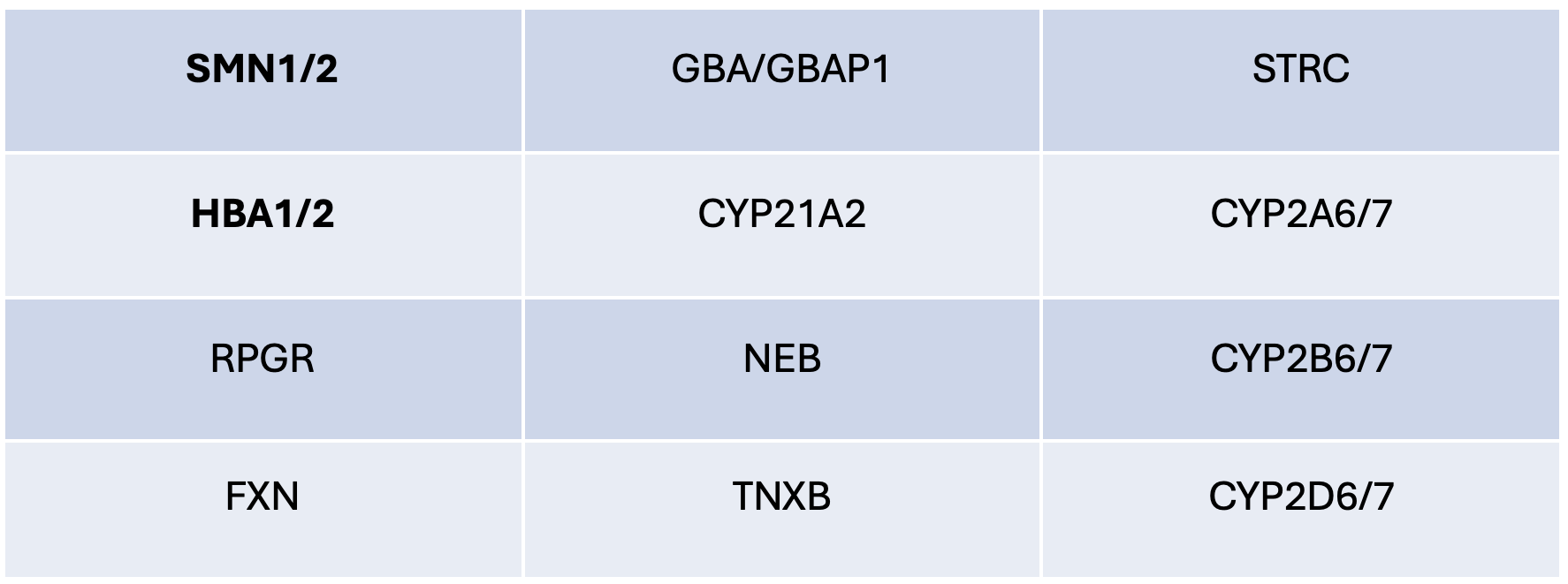
Table 1: List of genes with extended coverage in the spike-in panel
Targeted caller method overview
The DRAGEN HBA caller uses the sequencing depth signal over several unique subregions within and near the HBA locus (Figure 1A) to infer copy number genotype combinations common to that locus, thus overcoming the homology-mediated ambiguous read alignment issues in it. The DRAGEN HBA caller can detect copy number genotypes that cover a wide spectrum of molecular subtypes of α-thalassemia.
The DRAGEN SMN caller (Figure 1B) identifies the total copy number of SMN1 and SMN2 by counting and normalizing the number of reads aligned to either region. A separate copy number estimation is performed for regions spanning exons 1–6 and exons 7–8 to ensure appropriate handling of common exon 7–8 deletions. A scan is then performed across differentiating sites to determine the SMN1-specific copy number. SMN2 copy number is found by subtracting SMN1 copy number from total copy number.
The novel exome-based callers leverage the extended coverage provided by the spike-in probes, along with a panel of normal-based depth normalization (within batch), and the specialized CNV detection techniques of the original HBA and SMN callers to enable accurate HBA and SMN genotyping on exome data (Figure 1C).
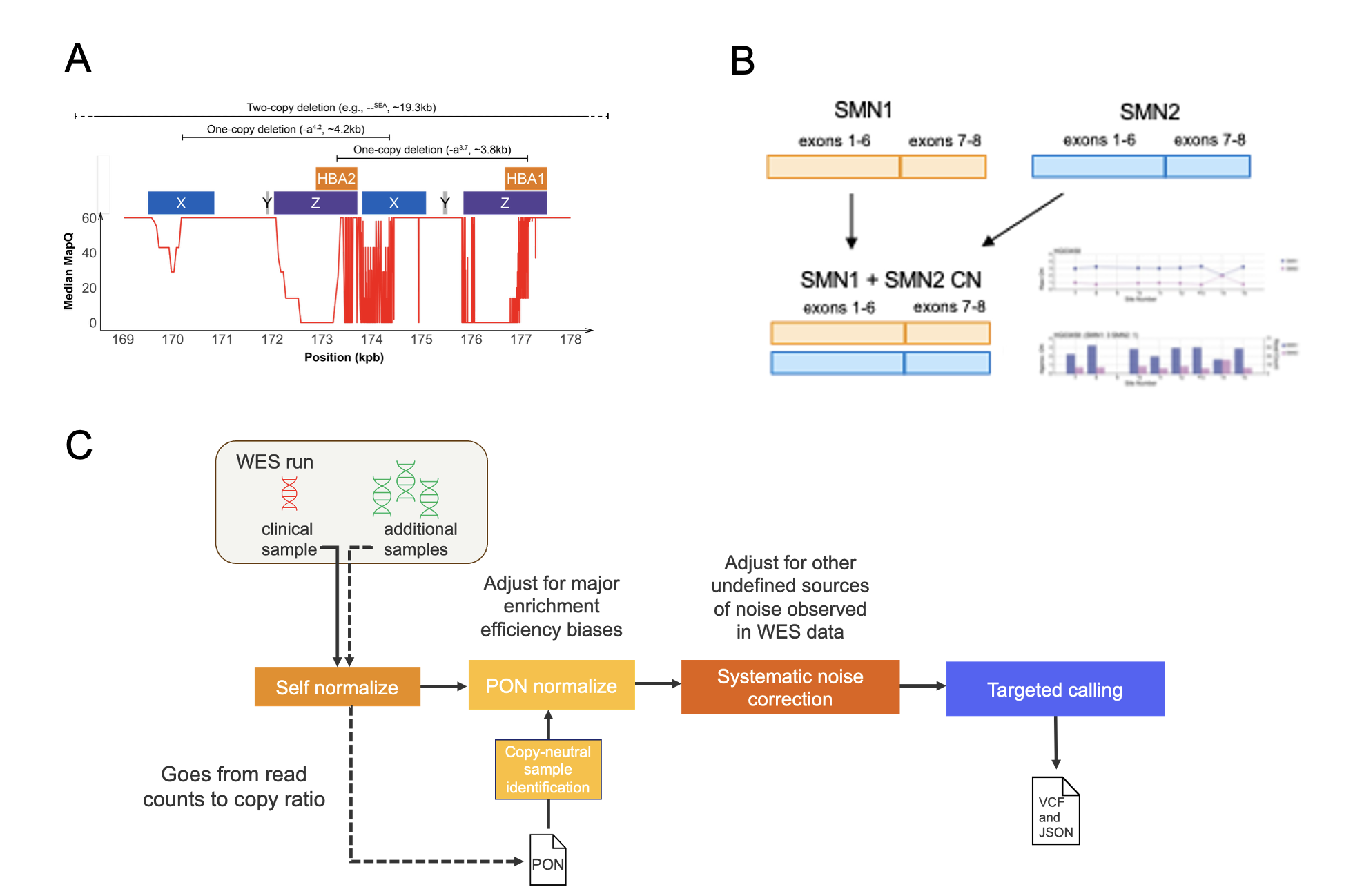
Figure 1A: Representation of the HBA1/2 locus features, median mapping quality of reads in that locus, and pathogenic copy number variants that frequently affect that locus. The HBA caller leverages unique subregions to infer copy number genotypes.
Figure1B: Schematic representation of the SMN1/2 copy number estimation workflow in the DRAGEN SMN caller
Figure1C: Schematic representation of the HBA and SMN caller workflows for WES datasets
Results on internal cell line cohort
We used cell lines profiled at Illumina with the exome panel and the added spike-in panel to assess accuracy during development of the assay. We evaluated SMN and HBA copy number genotype calls made from exome and spike-in data against orthogonally confirmed copy number genotypes for HBA (726 datasets from 203 unique samples) and SMN (739 datasets from 216 unique samples). The orthogonal results used in this study were derived either from DRAGEN targeted caller results on matched WGS data or from qPCR/MLPA, when available, from Coriell. Three different operators prepared all libraries manually and sequenced them on the NovaSeq 6000 System with 96 samples loaded on S4 flow cells.
HBA genotype calls derived from whole-exome sequencing (WES) using the updated targeted callers were 100% (710/710) concordant with orthogonal method calls in the cell line cohort with a 2.2% no-call rate. SMN1 copy number calls were 98.3% concordant with the orthogonal methods and 100% concordant in terms of SMA status classification (affected status defined as zero copies of SMN1; carrier status defined as one copy of SMN1; unaffected status defined as two or more copies of SMN1). The SMN2 copy number calls from WES were 99.3% concordant with the orthogonal methods. The no-call rate for SMN1/2 copy numbers in this cohort was 1.1%. We observed an overall QC failure rate of 5.9% across the cell line sample cohort, driven by samples with an overall depth profile that was highly divergent from other samples in the same batch due to cell line artifacts.
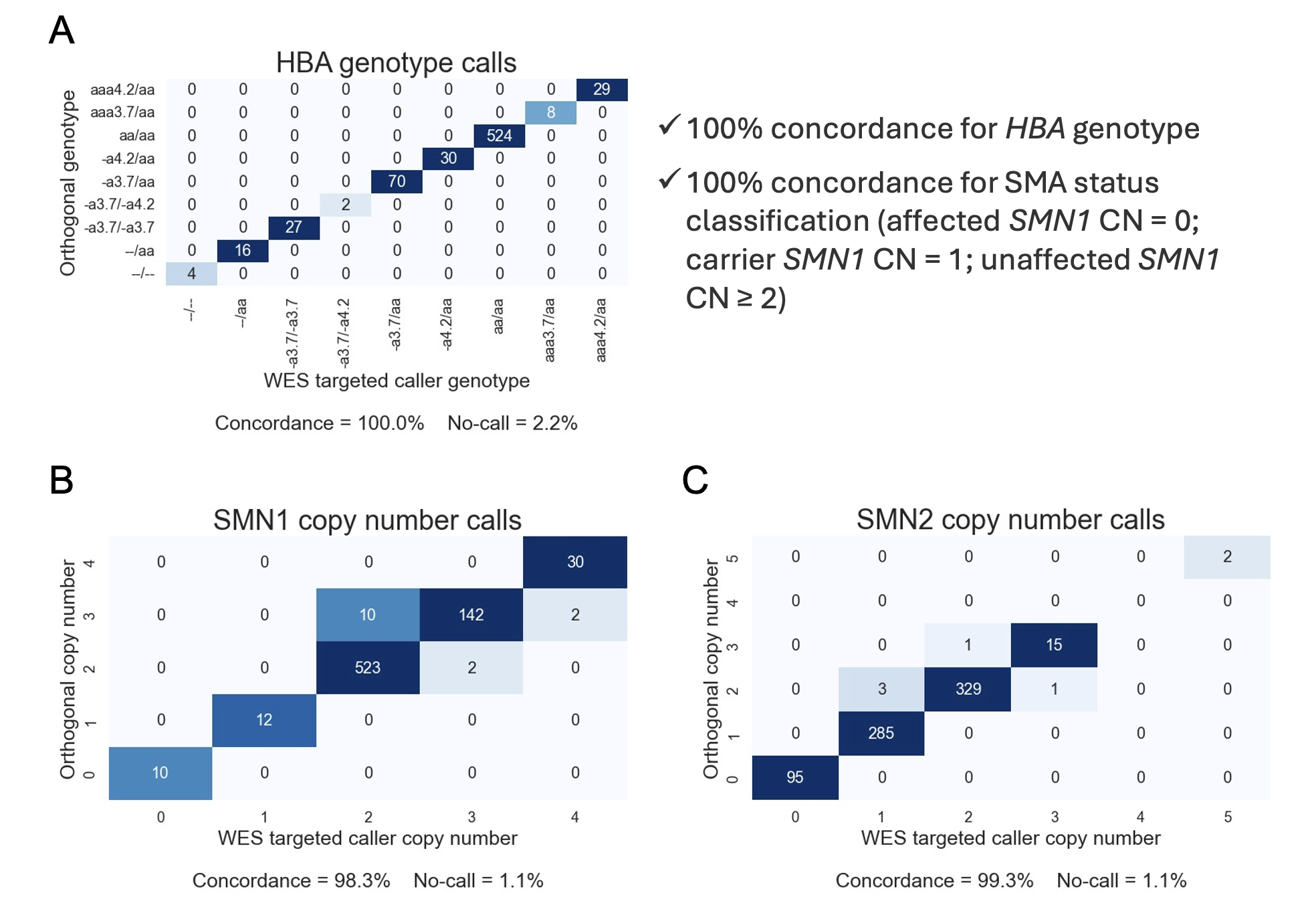
Figure 1A: Concordance analysis results between the WES-based HBA caller and the results from WGS-based HBA caller on matched datasets.
Figure2B: Concordance analysis results between SMN1 copy number calls obtained from the WES-based SMN caller and those from the WGS-based SMN caller on matched datasets.
Figure2C: Concordance analysis results between SMN2 copy number calls obtained from the WES-based SMN caller and those from the WGS-based SMN caller on matched datasets
Results on external clinical cohort
Juno Genetics in Spain generated an independent, partially blind (665/1117 blind for SMN and 464/1099 blind for HBA) and partially consecutive cohort of clinical samples profiled with Illumina DNA Prep with Exome 2.5 Enrichment with added Illumina CS/PGx Custom Enrichment Research Panel spike-in (1140 datasets from 1009 unique samples) and Illumina analyzed the cohort with DRAGEN v4.4. Juno prepared exome libraries using an automated system and sequenced them on the NovaSeq 6000 with either 192 samples loaded on each S4 flow cell or 96 samples loaded on S2 flow cells. Most samples in that cohort had matched results from MLPA profiling for HBA locus as well as qPCR or MLPA profiling of SMN1 copy number, which were used to evaluate the accuracy of the exome-based calls. The concordance between exome results with matched MLPA or qPCR tests among samples passing QC was 100% (1099/1099) for HBA and 99.8% for SMN1 copy number and SMA status (1113/1115). No samples failed QC due to discrepancies in depth profile against other samples in the batch in this cohort. The significantly higher percentage of samples passing QC between this clinical sample cohort (100%) and the cell line cohort Illumina used for development (94.1%) indicates that more robust and consistent results can be achieved on clinical samples prepared with automated systems than on cell lines prepared manually by different operators.
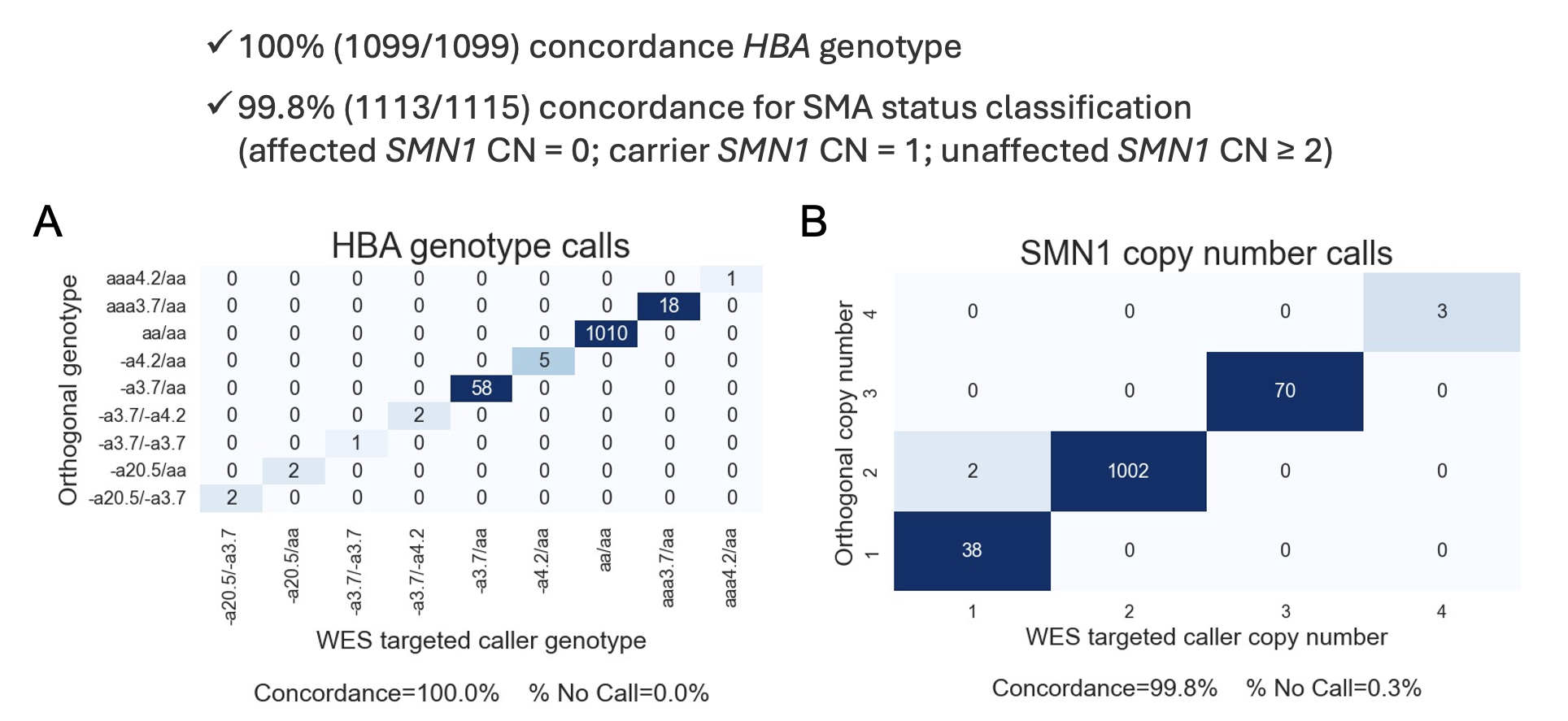
Figure 1A: Concordance analysis results between the WES-based HBA caller and orthogonal assay (MLPA) results on the clinical sample cohort.
Figure3B: Concordance analysis results between SMN1 copy number calls obtained from the WES-based SMN caller and orthogonal assay (qPCR) results on the clinical sample cohort.
Conclusion
The novel exome-based HBA and SMN targeted callers implemented in DRAGEN and Illumina CS/PGx Custom Enrichment Research Panel added to the Illumina DNA Prep with Exome 2.5 Enrichment demonstrate a reliable approach for profiling the HBA and SMN genes within an integrated NGS workflow. This method has the potential to reduce reliance on orthogonal assays in carrier screening research, enhancing cost-effectiveness and improving the accessibility of such data sets.