Summary
- α-thalassemia is caused by mutations in high homology HBA1 and HBA2 genes (~97%).
- We present here a new WGS-based DRAGEN HBA caller that can accurately detect deletional and non-deletional variants in the HBA locus.
- We show high copy number genotype concordance between the DRAGEN HBA caller and orthogonal long read technology.
- We also applied the DRAGEN HBA caller on a large sample cohort with diverse ancestry. The caller can call copy number genotypes for the vast majority of samples and achieves high Mendelian consistency on trios.
- This population screening analysis suggests that African and South Asian superpopulations have high carrier frequencies of α-thalassemia, and that two-copy deletion in cis has highest frequency in the Dai Chinese population, which are in line with previous findings.
- With this tool, large scale population genomics studies utilizing WGS can now investigate the distribution of variants in the HBA locus and help guide decisions about how to best deploy carrier and newborn screening tests.
α-thalassemia is caused by mutations in HBA1 and HBA2 genes
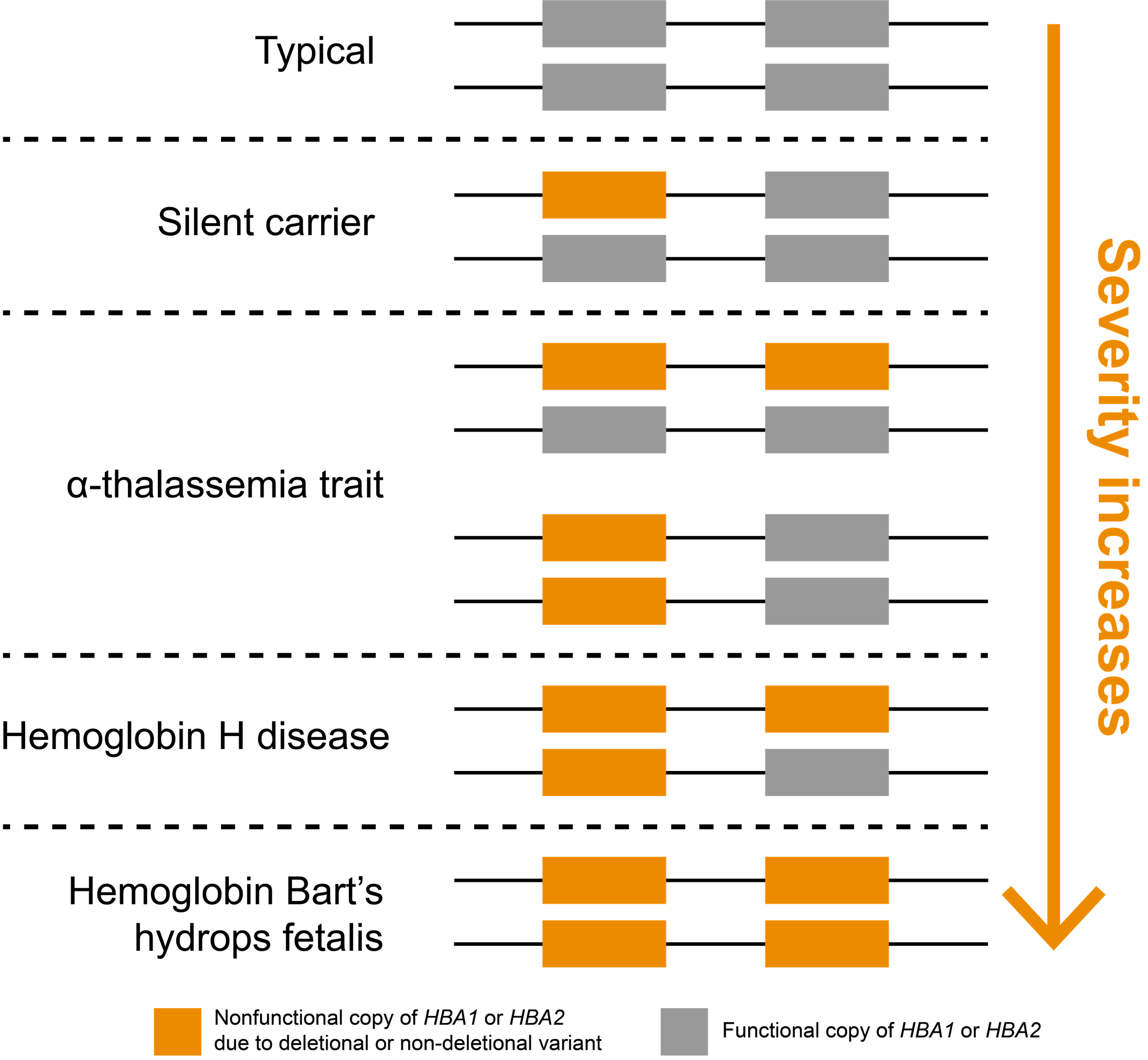
In humans, the two genes that encode α-hemoglobin, HBA1 and HBA2, are located back-to-back on chromosome 16, meaning that most people have four total gene copies that produce α-hemoglobin chains (Figure 1). Inheriting at least two mutated copies of either HBA1 or HBA2 genes leads to α-thalassemia, an autosomal recessive blood disorder. Importantly, loss of one or more of these four copies is very common in some parts of the world. It is believed this may be driven evolutionarily by heterozygous carriers, who are less susceptible to malarial parasites.7
Reproductive carrier and newborn screening of α-thalassemia is recommended due to its high carrier frequency
Due to the frequent loss of HBA1 or HBA2, α-thalassemia has become one of the most common human monogenic disorders worldwide, with more than 300,000 severely affected individuals worldwide.9 This means that more than 4% of the world’s population are carriers of pathogenic variants in HBA1 or HBA2.10 In certain regions, such as sub-Saharan Africa and South Asia, the carrier frequency can exceed 30%.10 Due to its high carrier frequency and the severity of associated clinical effects, population-wide carrier screening for α-thalassemia is recommended by the American College of Medical Genetics and Genomics6 (tier 3 list) and the American College of Obstetricians and Gynecologists4 for all women who are pregnant or are planning a pregnancy. Newborn screening for α-thalassemia is also recommended by the Centers for Disease Control and Prevention.2 Given the wide variability in prevalence of α-thalassemia and the uneven distribution of the variants responsible for the phenotype in different subpopulations, a complete understanding of the population specific distribution of pathogenic HBA1 and HBA2 variants is key to help guide the deployment of newborn and carrier screening tests for this condition.
α-thalassemia severity is positively correlated with the number of nonfunctional copies of HBA1 and HBA2 genes
Noncarrier individuals have a combined total of four functional α-globin genes: two each of HBA1 and HBA2. In affected individuals, symptom severity is correlated with the number of nonfunctional copies (Figure 1). Whole-gene deletions (“deletional”) are the most common type of loss-of-function variants for these genes, though other types of variants may also be pathogenic (“non-deletional”). Individuals carrying one nonfunctional copy of either HBA1 or HBA2, thus having three functional copies, are typically asymptomatic and are referred to as “silent carriers.” Individuals carrying two nonfunctional copies have “α-thalassemia trait” (or “α-thalassemia minor”). These individuals are carriers who might have non-severe symptoms, such as mild anemia. The configuration of their two nonfunctional copies (cis or trans) does not impact symptoms but can have implications for reproduction.
Three nonfunctional copies of HBA1 or HBA2 is associated with hemoglobin H (HbH) disease, which can cause moderate to severe anemia depending on the underlying variant types. If all four copies of HBA1 and HBA2 genes are nonfunctional, a person has “α-thalassemia major,” otherwise known as “hemoglobin Bart’s hydrops fetalis,” which is the most severe type of α-thalassemia. Most individuals with this condition die prenatally or soon after birth. It could also lead to increased risk of complications for the mother (e.g., preeclampsia).
A novel WGS-based variant calling method for the HBA locus
Traditionally, molecular testing of α-thalassemia is done using Sanger sequencing or multiplex ligation-dependent probe amplification on PCR-enriched α-globin gene fragments. More recently, targeted NGS technology has been demonstrated as a screening and confirmation method for α-thalassemia with high sensitivity.8 These enrichment-based assays require extra labor and longer preparation time compared to WGS-based assay.
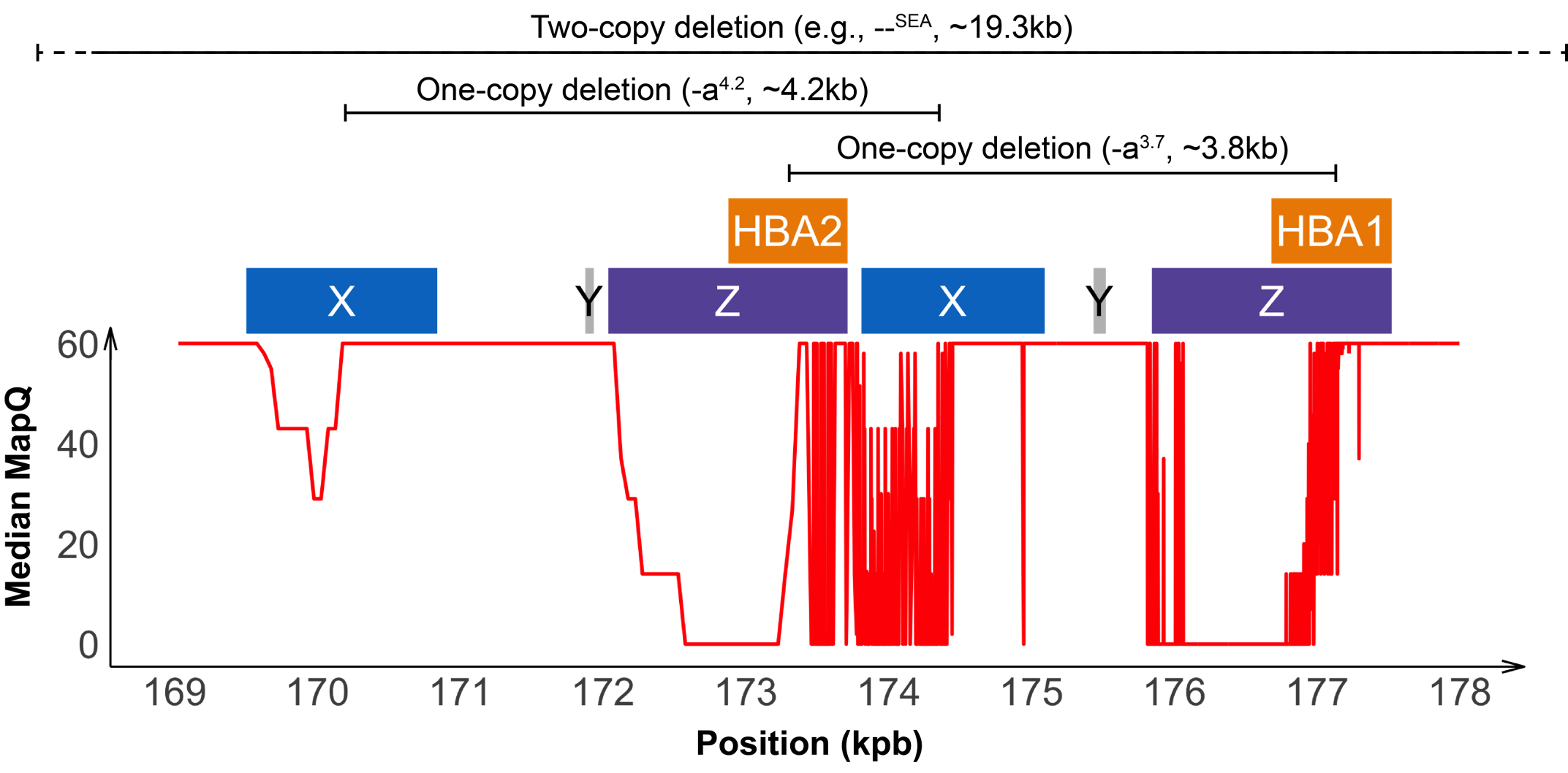
Detecting variants in the HBA locus can be challenging, in part due to high homology between HBA1 and HBA2 genes (~97%) and their nearby regions, resulting in ambiguous read alignments (Figure 2). The duplicated HBA1 and HBA2 genes are within two highly homologous 4 kilobase units, each consisting of three “homologous boxes” (Figure 2) that play a part in the common deletional types of α-thalassemia. Z and X box mispairing can lead to unequal homologous recombination during meiosis, giving rise to the -α3.7 (~3.8 kb) and -α4.2 (~4.2 kb) deletions, respectively, which are the most common deletional variants related to α-thalassemia. Other common deletional variants include the two-copy deletions in cis that remove both HBA1 and HBA2 genes in one chromosome. The most common form of two-copy deletion is the --SEA deletion that is prevalent in the Southeast Asian population.5 Overall, the high homology regions at the HBA locus create challenges for identifying variants that reside within this locus, demanding a purpose-built informatics solution.
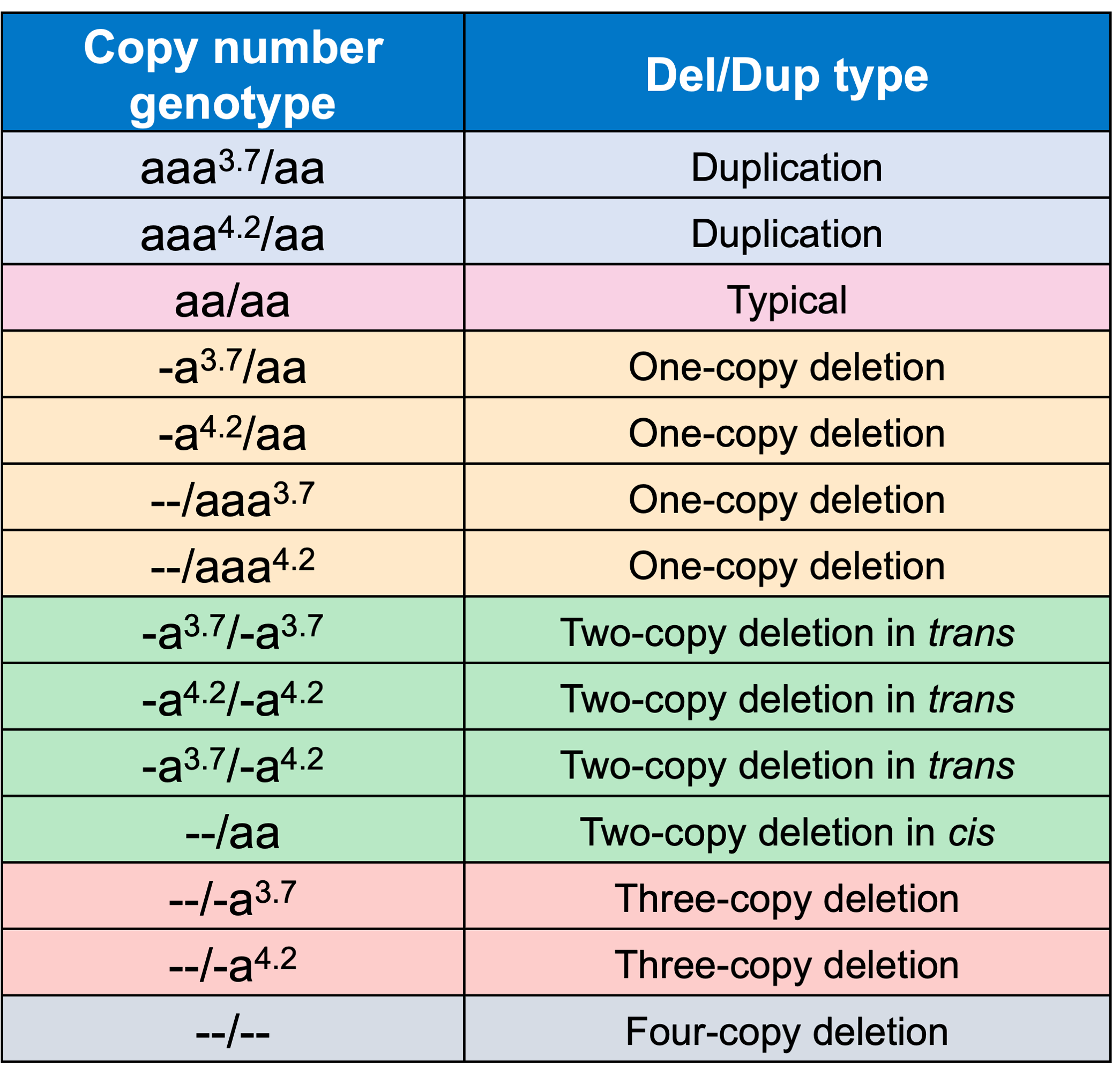
About 95% of α-thalassemia cases result from gene deletional rather than non-deletional variants.11 However, non-deletional variants have been reported to result in more severe phenotypes.12 The DRAGEN HBA caller also covers 17 small variants that have been reported in ClinVar as pathogenic or likely pathogenic by multiple laboratories.3 The ability to call HBA1 and HBA2 variants through WGS enables population sequencing efforts as well as α-thalassemia research projects to gain insights into this important locus.
High copy number genotype concordance between the DRAGEN HBA caller and orthogonal long read technology
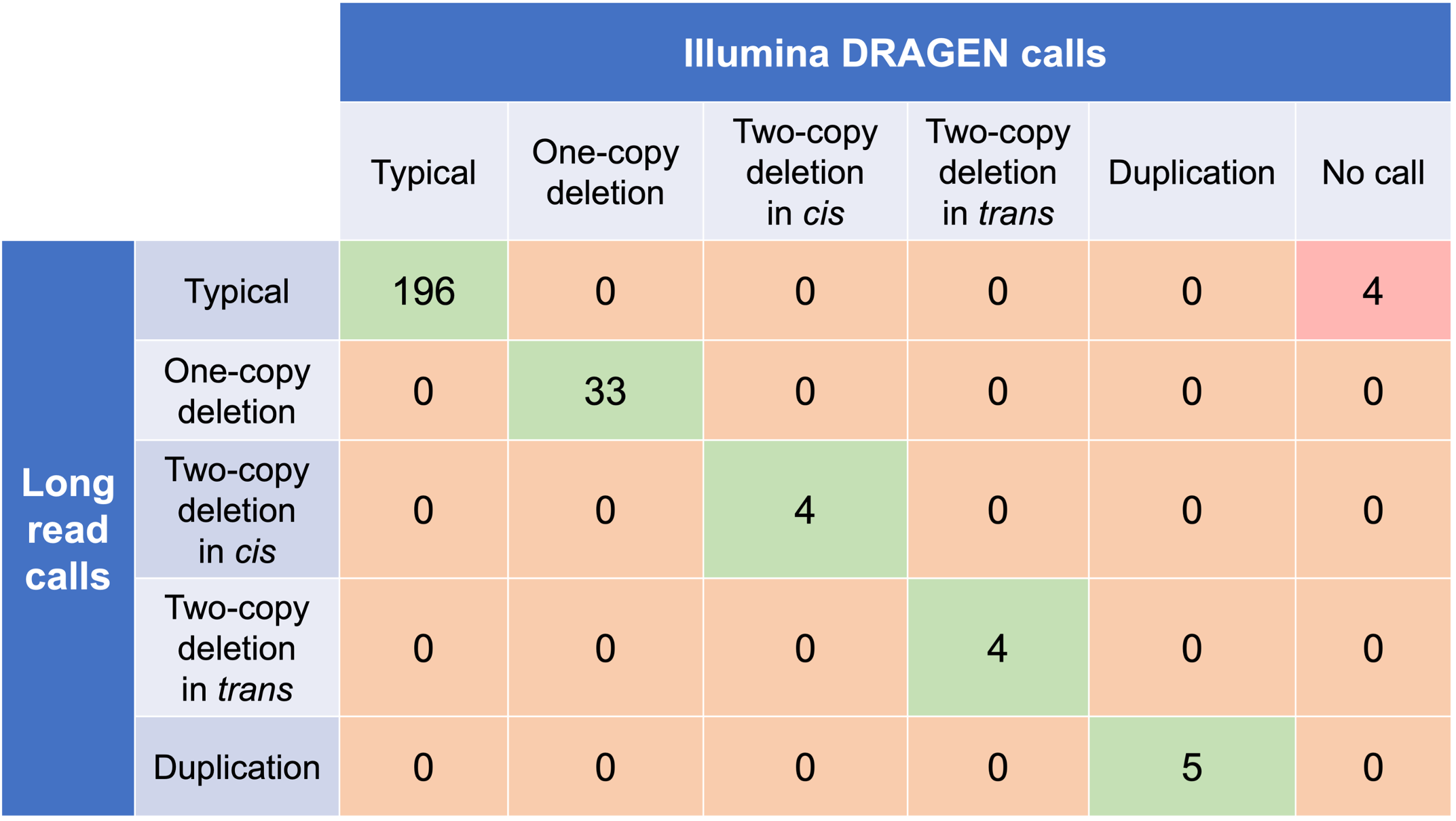
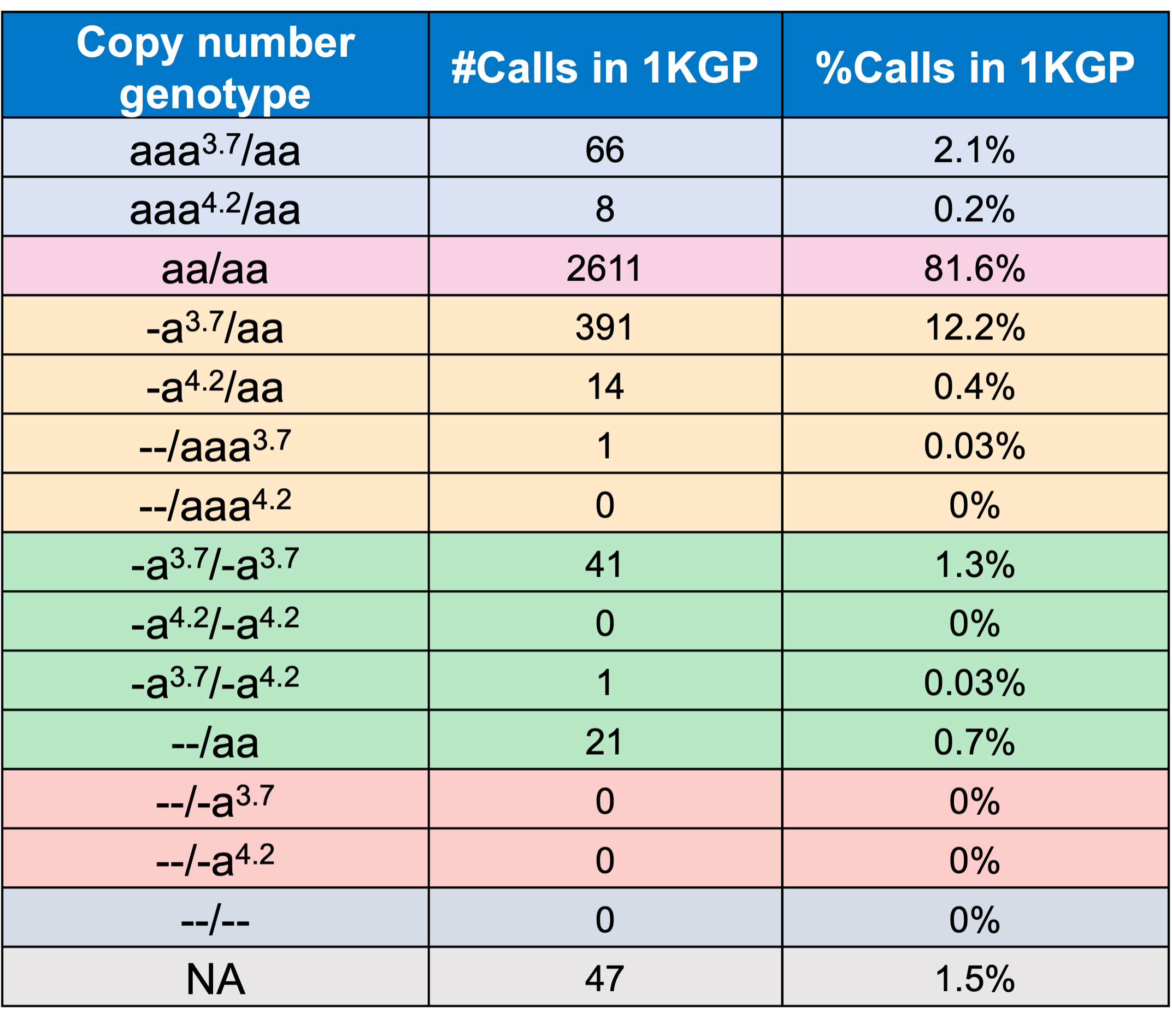
In order to evaluate the DRAGEN HBA caller, we first compared the copy number genotypes from our caller to those from an orthogonal long read technology (PacBio HiFi). The copy number genotypes from long read data were made based on the alignment pileups, phased assemblies, and SV calls. Using 246 samples of diverse genetic backgrounds from the 1000 Genomes Project (1KGP) with matching short read and long read sequencing data, the DRAGEN HBA caller performed with high concordance with orthogonal long read technology for all major copy number genotypes (Table 2). The no call rate from the DRAGEN HBA caller was 1.6% (4/246). Future work could include evaluation of the sources for the 4 no call cases.
The DRAGEN HBA caller shows high trio Mendelian consistency
We next evaluated the Mendelian transmission of copy number genotype calls from the DRAGEN HBA caller on the trio data set from 1KGP. This test checks whether the copy number genotype in an individual could have been received from their biological parents based on Mendelian inheritance. In 575 trios from 1KGP that we have copy number genotype calls for, 100% of trio calls are consistent with Mendelian inheritance. This result suggests that the DRAGEN HBA caller achieves consistent genotypes across the pedigree but does not guarantee the accuracy of the copy number genotypes called. Combining the trio Mendelian transmission test with the orthogonal technology copy number genotype concordance test provides confidence in the overall accuracy and reliability of the copy number genotypes produced by the DRAGEN HBA caller.
The DRAGEN HBA caller found population-level genotypes consistent with prior studies
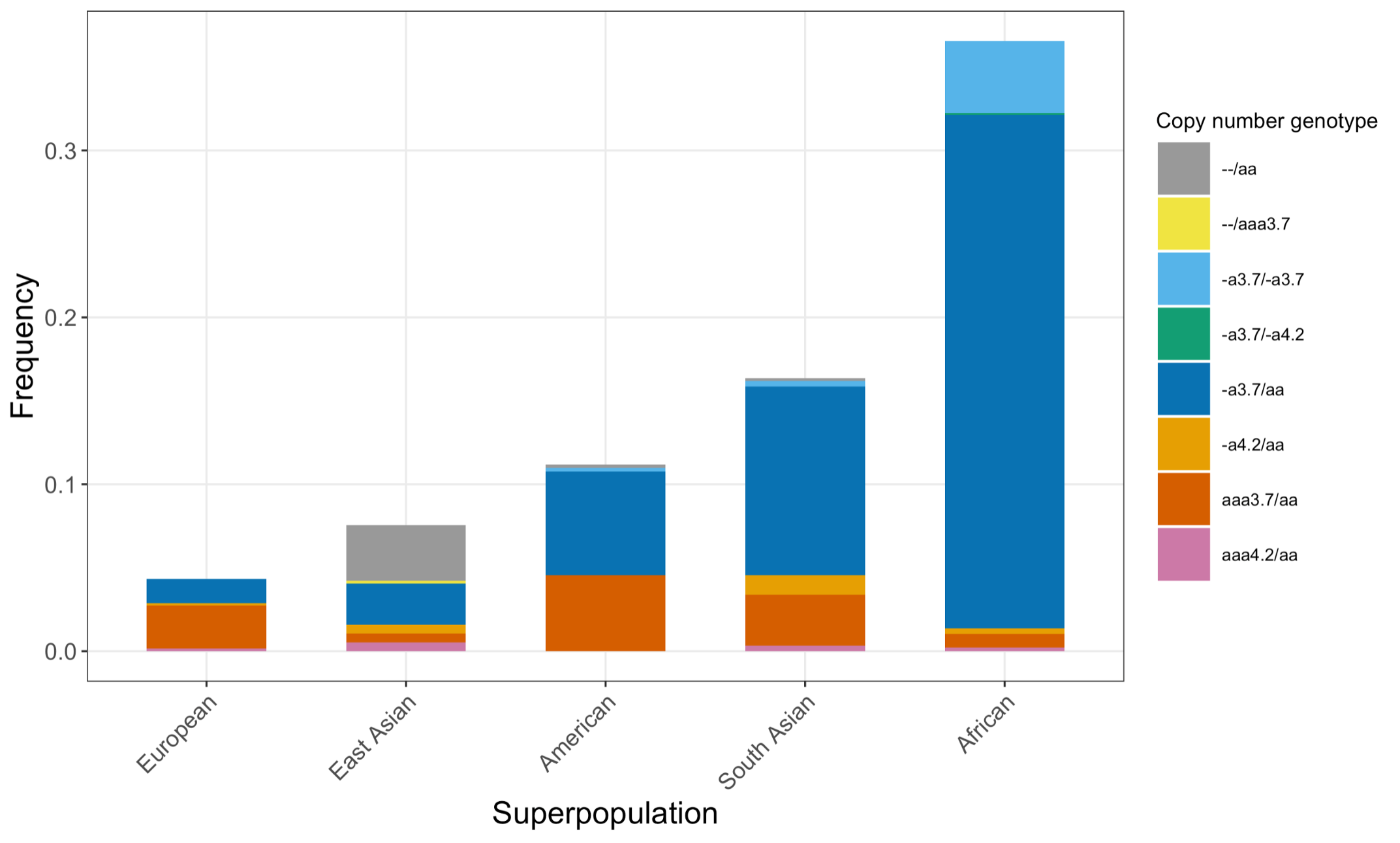
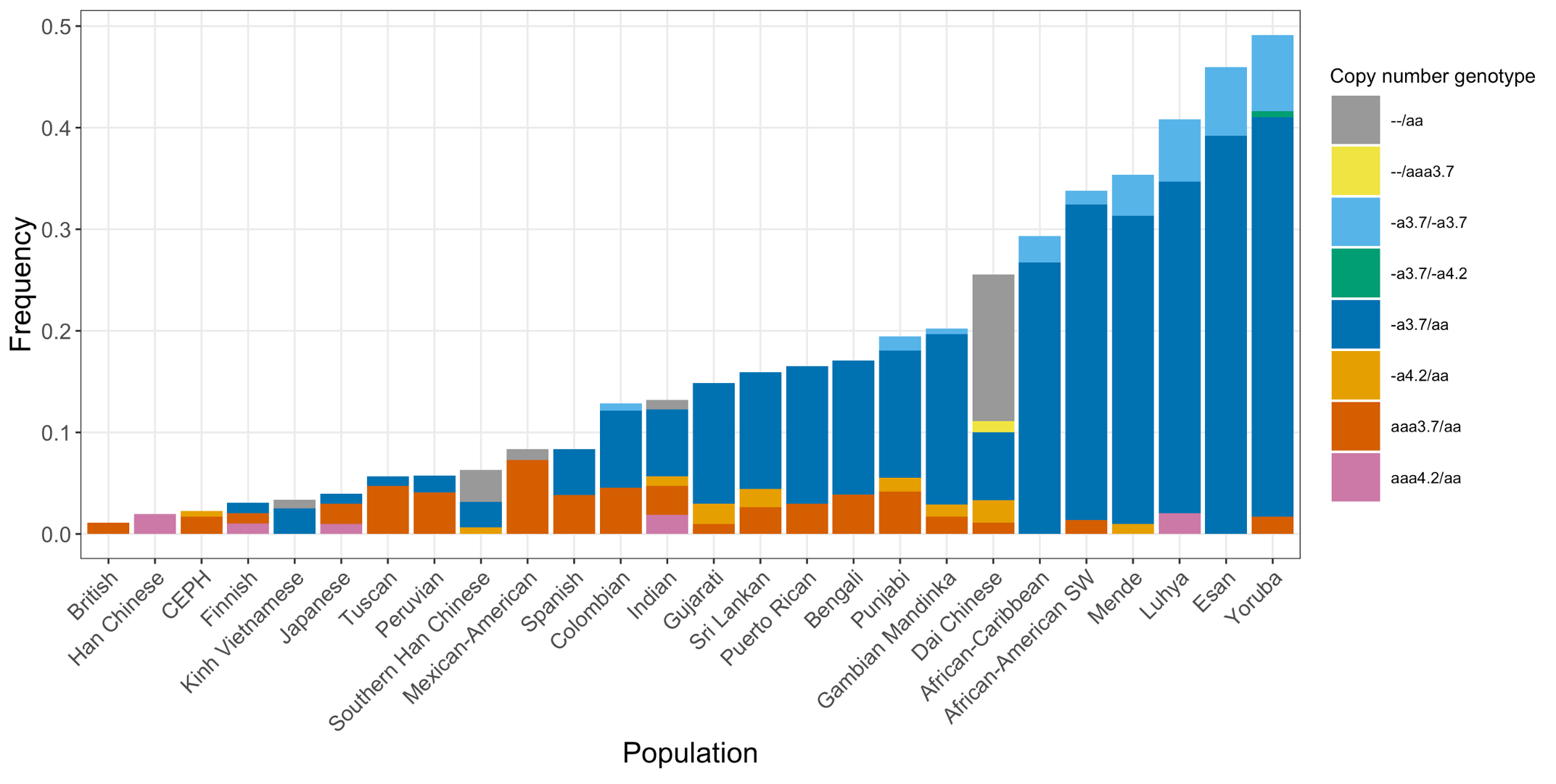
Finally, we applied the DRAGEN HBA caller on an extended data set of 3201 samples with diverse genetic background from 1KGP. The DRAGEN HBA caller was able to call copy number genotypes for 98.5% of samples (Table 3), confirming the low no call rate observed in the smaller data set shown previously. The carrier frequencies in the African and South Asian populations are approximately 36% (313/881) and 13% (77/593) (Figure 3), respectively, and the two-copy deletion in cis has the highest frequency in the Dai Chinese population (~14%, 13/90) (Figure 4). This is consistent with previous findings.8 Additionally, the DRAGEN HBA caller detected five samples carrying one pathogenic/likely pathogenic small variant in the 1KGP data set (Table 4). The low frequency of small variants discovered in this data set is also consistent with previous findings.11

Availability
The HBA caller will be available in the 4.2 release of DRAGEN. Please contact ffg-info@illumina.com to request access to the DRAGEN HBA caller.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
Acknowledgment
We thank Mitch Bekritsky, Severine Catreux, James Han, Carri-Lyn Mead, and Sam Strom at Illumina for providing comments on this article.
December 14, 2022: This article has been updated to reflect a revision to the orthogonal data set used to evaluate the DRAGEN HBA caller. The revised data set, with a few errors corrected, cleared two discordant cases between the DRAGEN HBA caller and PacBio HiFi reads.
References
1. Achour, Ahlem, Tamara T. Koopmann, Frank Baas, and Cornelis L. Harteveld. “The Evolving Role of Next-Generation Sequencing in Screening and Diagnosis of Hemoglobinopathies.” Frontiers in Physiology 12 (July 27, 2021): 686689. https://doi.org/10.3389/fphys.2021.686689.
2. Bender, M. A. “Newborn Screening Practices and Alpha-Thalassemia Detection — United States, 2016.” MMWR. Morbidity and Mortality Weekly Report 69 (2020). https://doi.org/10.15585/mmwr.mm6936a7.
3. ClinVar Variation ID: 280127, 375746, 15624, 15627, 15647, 15652, 15656, 15662, 15687, 15690, 15849, 375749, 439126, 439112, 618674, 801169, and 811900 (https://www.ncbi.nlm.nih.gov/clinvar/)
4. “Carrier Screening for Genetic Conditions | ACOG.” Accessed August 1, 2022. https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2017/03/carrier-screening-for-genetic-conditions.
5. Fucharoen, S., and P. Winichagoon. “Thalassemia in SouthEast Asia: Problems and Strategy for Prevention and Control.” The Southeast Asian Journal of Tropical Medicine and Public Health 23, no. 4 (December 1992): 647–55.
6. Gregg, Anthony R., Mahmoud Aarabi, Susan Klugman, Natalia T. Leach, Michael T. Bashford, Tamar Goldwaser, Emily Chen, et al. “Screening for Autosomal Recessive and X-Linked Conditions during Pregnancy and Preconception: A Practice Resource of the American College of Medical Genetics and Genomics (ACMG).” Genetics in Medicine 23, no. 10 (October 1, 2021): 1793–1806. https://doi.org/10.1038/s41436-021-01203-z.
7. Harteveld, Cornelis L, and Douglas R Higgs. “α-Thalassaemia.” Orphanet Journal of Rare Diseases 5 (May 28, 2010): 13. https://doi.org/10.1186/1750-1172-5-13.
8. He, Jing, Wenhui Song, Jinlong Yang, Sen Lu, Yuan Yuan, Junfu Guo, Jie Zhang, et al. “Next-Generation Sequencing Improves Thalassemia Carrier Screening among Premarital Adults in a High Prevalence Population: The Dai Nationality, China.” Genetics in Medicine 19, no. 9 (September 1, 2017): 1022–31. https://doi.org/10.1038/gim.2016.218.
9. Higgs, Douglas R. “The Molecular Basis of α-Thalassemia.” Cold Spring Harbor Perspectives in Medicine 3, no. 1 (January 2013): a011718. https://doi.org/10.1101/cshperspect.a011718.
10. Piel, Frédéric B., and David J. Weatherall. “The α-Thalassemias.” Review-article. http://dx.doi.org/10.1056/NEJMra1404415. Massachusetts Medical Society, November 12, 2014. World. https://doi.org/10.1056/NEJMra1404415.
11. Pornprasert, Sakorn, Nur-afsan Salaeh, Monthathip Tookjai, Manoo Punyamung, Panida Pongpunyayuen, and Kallayanee Treesuwan. “Hematological Analysis in Thai Samples With Deletional and Nondeletional HbH Diseases.” Laboratory Medicine 49, no. 2 (March 21, 2018): 154–59. https://doi.org/10.1093/labmed/lmx068.
12. Sabath, Daniel E. “Molecular Diagnosis of Thalassemias and Hemoglobinopathies: An ACLPS Critical Review.” American Journal of Clinical Pathology 148, no. 1 (July 1, 2017): 6–15. https://doi.org/10.1093/ajcp/aqx047.