Introduction
Short tandem repeats (STRs) are found throughout the genome and present in the exons of most (92%) human genes.1 While the biological functions of most STRs are unknown,2 there is evidence linking some STRs to human disease1 in a class of genetic conditions known collectively as repeat expansion (RE) disorders. To date, over 40 neurogenetic conditions have been identified as RE disorders, including Huntington’s disease, amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Fragile X syndrome.3 The ability to identify clinically significant REs across the genome is critical for both diagnosing RE disorders and to provide a comprehensive assessment of medically relevant regions of the genome. Current measures for RE assessment include targeted methods, such as PCR-based or Southern blot assays,6 which can be costly and time consuming.
PCR-free whole-genome sequencing (WGS) can provide a nearly unbiased survey of the entire genome. Many clinical laboratories have already implemented WGS as a diagnostic tool for identifying small variants such as single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) and copy number variants (CNVs), but not for more complex structural variants (SVs) such as STR expansions. In the case of RE assessment, a lack of both the tools to accurately call expanded STR variants and the evidence of analytical validity regarding the performance of tools has been a primary limitation to clinical implementation.
Recent advances in algorithms designed specifically for genotyping STRs,7,8 such as the ExpansionHunter software package, have shown that WGS can provide reliable detection of REs across the genome with high sensitivity and high specificity.9 WGS combined with ExpansionHunter has revealed previously undiagnosed neurological RE disorders, including in patients with no prior family history or suspicion of a RE disorder.9 The addition of a visual inspection step to analyze the read-level evidence that supports a genotype call by a trained professional, as recommended by the Association for Medical Pathology (AMP) and the College of American Pathologists (CAP),10 provides further support for the accuracy of a given call during analysis. For small variants, visual inspection is commonly performed by analyzing alignments using the Integrated Genome Viewer (IGV),11 but complex variants that differ significantly from the reference genome, such as REs, require visualization tools specifically designed for these variants. In this article, we describe the validation and implementation of WGS to detect pathogenic REs and illustrate how the addition of a software-enabled visual inspection step to the clinical pipeline has significantly reduced the number of false-positive results.
Calling REs with ExpansionHunter
ExpansionHunter estimates repeat sizes from WGS data at a given locus through the creation of a dynamic graph reference genome where the repeat is represented by a loop in the graph and sequence reads are then aligned to this dynamic graph.8 If the repeat is significantly shorter than the read length of the sequence data (eg, 150 bp), there should be spanning reads available to use to identify the exact size of the repeat. If the length of the repeat is longer than the read length, then reads that are fully within the repeats (in-repeat reads, or IRRs) can be identified and counted to provide a probabilistic estimate of the size of the repeat.
Data visualization with REViewer
Laboratory practices include use of tools such IGV for visual data inspection. However, these tools rely on aligning data to a standard reference genome, which can be problematic for larger REs that may include insertions of a significant amount of additional sequence. To address the gap between IGV software and RE investigation, Illumina developed Repeat Expansion Viewer (REViewer), a software tool that creates a static visualization of the WGS reads containing the repeat identified by ExpansionHunter.12 Within REViewer, reads are aligned to the two haplotypes represented by the genotype identified in ExpansionHunter and the read pileups are plotted as static images. The resulting plots enable direct visualization of the haplotypes and the corresponding read pileup of the ExpansionHunter genotypes (Figure 1). From the visualization, users can identify and interrogate sequencing reads that align to the region, allowing for an additional assessment of the ExpansionHunter calls, analogous to how IGV is used to visually confirm small variant calls. Based on this visualization it is possible to review the genotype calls made by ExpansionHunter to identify putative false positives or protective interruptions (ie, AGG interruptions in FMR1), which may allow the laboratory to conduct orthogonal investigation of the variant if relevant for the indication for testing.
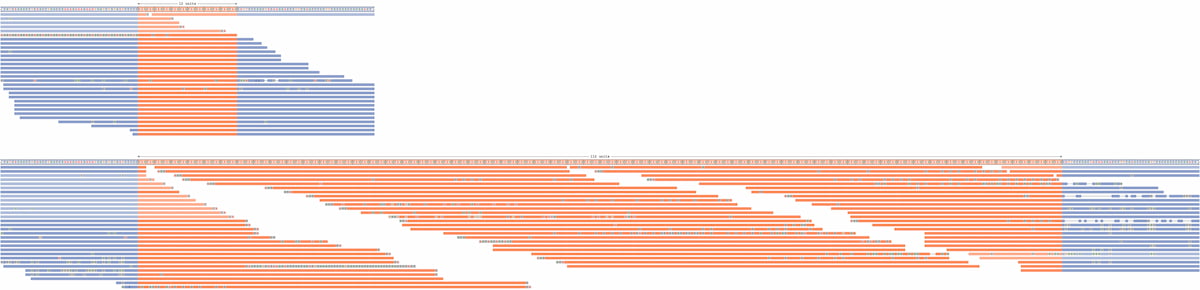
Figure 1: Visualization of detected REs using ExpansionHunter—A read pileup for a DMPK repeat with an expansion on one allele. REViewer distributed the reads throughout the repeat to achieve similar coverage across the entire haplotype, supporting the presence of an expanded repeat. Note that alignment positions of reads within the repeat are chosen randomly. Alignments depicted in fainter colors correspond to reads that can be assigned to either allele.
WGS pipeline increases identification of ERs
To validate the use of ExpansionHunter and REViewer to identify REs in WGS data, scientists from the Genomics England Project and Illumina Clinical Services Laboratory (ICSL) employed a WGS workflow to retrospectively assess 13 of the most common genetic neurological RE disorders in 793 previously tested samples.9 Comparing the ExpansionHunter output against this benchmark data set showed correct classification of 215 out of 221 expanded alleles and 1316 out of 1321 normal alleles test. This indicated a total sensitivity of 97.3% (95% Confidence Interval (CI): 94.2%-99%) and specificity of 99.6% (95% CI: 99.1%-99.9%). All calls were visually inspected and reclassified as appropriate based on the quality of the reads supporting each call.[*] Following visual inspection and correction[PD1] , sensitivity was 99.1% (95% CI: 96.7%-99.9%) and specificity of 100% (95% CI: 99.7%-100%) (Table 1). Visualization of the expanded calls was able to detect false positives and reclassify all false negative alleles in one of the genes where only one allele was classified as expanded in samples with biallelic expansions. These results provide support for establishing this WGS pipeline in a clinical laboratory for identifying REs at a particular locus of interest classified as either “normal” or “expanded”.
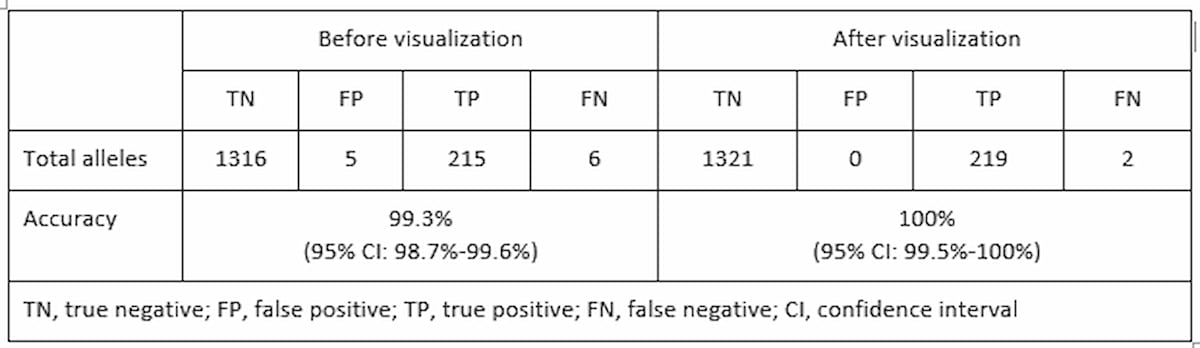
Table 1: Increased accuracy with visual inspection—Performance based on total number of normal and expanded alleles across all loci tested after visual inspection.9
Examples of WGS pipeline use for RE diagnosis
The Illumina Clinical Services Laboratory (ICSL) conducted a clinical validation of the ExpansionHunter software and implemented ExpansionHunter and a visual inspection workflow (Figure 2) in December 2019 as part of a clinical WGS test for patients with rare and undiagnosed genetic disease. The ICSL test definition includes review of 21 clinically relevant RE disorders as part of the Illumina iHope Program, an Illumina philanthropic initiative that aims to provide clinical WGS to individuals with limited access to molecular testing. To date, ICSL has issued seven clinical reports with clinically significant RE disorders, including myotonic dystrophy (DMPK), spinocerebellar ataxia type 7 (ATXN7), spinocerebellar ataxia type 8 (ATXN8OS) and Friedreich ataxia (biallelic expansions in FXN).
In one case, through the iHope Program from the Democratic Republic of Congo in collaboration with the University of Kinshasa, ICSL received samples from three adult sisters presenting to the clinic with spinocerebellar ataxia, pontocerebellar atrophy, skeletal muscle atrophy, abnormal pyramidal signs, extrapyramidal muscular rigidity, hypertonia, dysarthria, partial blindness, and a paternal family history of similar features. Clinical WGS analysis and ExpansionHunter identified an expansion of a CAG trinucleotide repeat in the ATXN7 gene associated with spinocerebellar ataxia type 7. The phenotypic spectrum of spinocerebellar ataxia type 7 includes adolescent or adult-onset progressive cerebellar ataxia and visual manifestations, matching the clinical features described in the three sisters. A clinical report describing these findings was issued to the family’s physician.
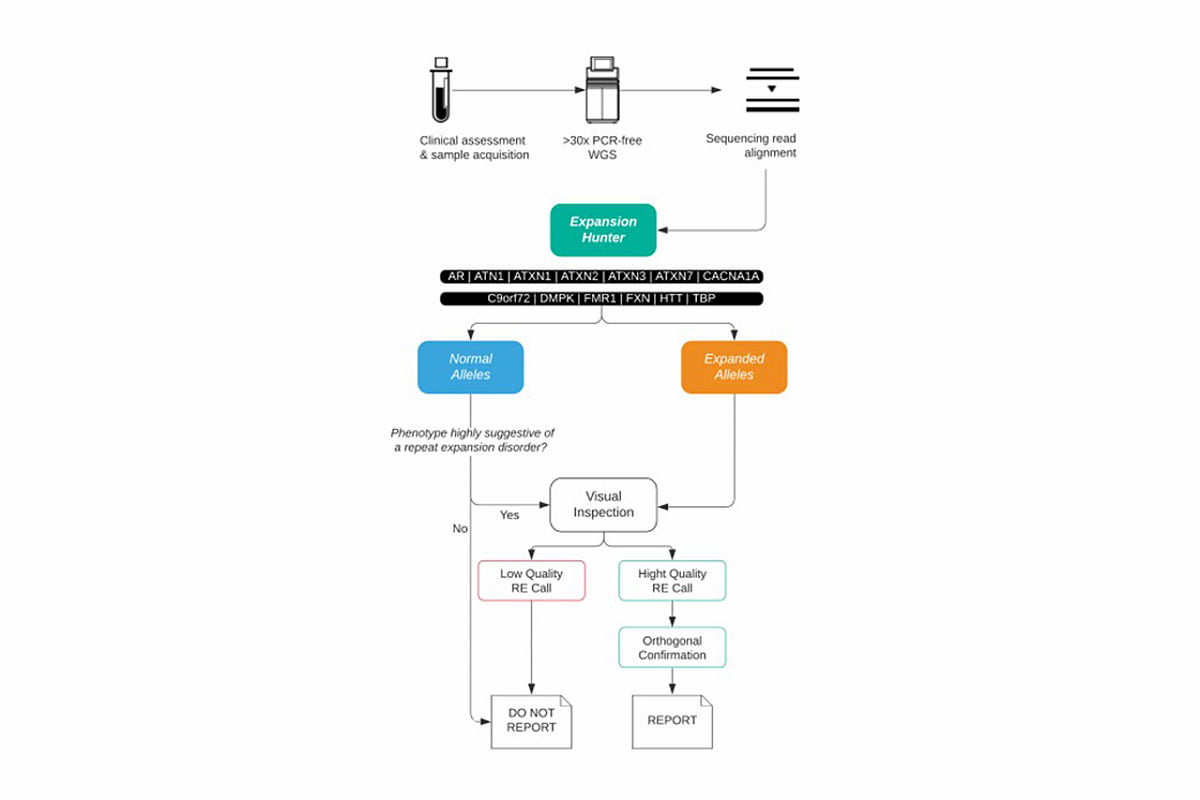
Figure 2: Suggested clinical workflow for use of WGS as a diagnostic tool for REs—After initial clinical assessment, WGS data is analyzed using ExpansionHunter. If an expansion is called or the phenotype might be associated with an RE disorder, a visual inspection of the reads considered by ExpansionHunter is recommended. If the quality of the reads appears to be high, the sample will be sent for orthogonal testing to confirm and characterize the length of the repeat. If the expansion looks to be a poor call, only those patients with strong phenotypic overlap may be sent for both orthogonal confirmation and characterization.
Discussion
Here we describe the utility of a clinical pipeline that uses WGS, ExpansionHunter, and REViewer to identify and visually inspect RE disorders of clinical interest. This pipeline is currently in use in multiple clinical laboratories and the ICSL team uses this pipeline to issue clinical reports with validated REs identified. Both ExpansionHunter and REViewer are freely available for download from github.com/illumina/ExpansionHunter and github.com/illumina/REViewer, respectively.
Learn more
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
Related content
Rare disease genomics, illumina.com/areas-of-interest/genetic-disease/rare-disease-genomics
Genetic disease testing, illumina.com/areas-of-interest/genetic-disease
Whole-genome sequencing, illumina.com/techniques/sequencing/dna-sequencing/whole-genome-sequencing
References
- Madsen BE, Villesen P, Wiuf C. Short tandem repeats in human exons: a target for disease mutations. BMC Genomics. 2008;9:410. Published 2008 Sep 12. doi:10.1186/1471-2164-9-410
- Fan H, Chu JY. A brief review of short tandem repeat mutation. 2007;5(1):7-14. doi:10.1016/S1672-0229(07)60009-6
- Paulson HL. Chapter 9-Repeat expansion diseases. In: Geschwind DH, Paulson HL, Klein C, editors. Handbook of Clinical Neurology (Neurogenetics, Part I; vol. 147) [Internet]. Elsevier; 2018:105–123. Available at: http://www.sciencedirect.com/science/article/pii/B9780444632333000099. Accessed October 14, 2020.
- Gossye H, Engelborghs S, Van Broeckhoven C, van der Zee J. C9orf72 Frontotemporal Dementia and/or Amyotrophic Lateral Sclerosis. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; January 8, 2015. Accessed October 14, 2020. Clinical neurogenetics: autosomal dominant spinocerebellar ataxia.
- Shakkottai VG, Fogel BL. Clinical neurogenetics: autosomal dominant spinocerebellar ataxia. Neurol Clin. 2013;31(4):987-1007. doi:10.1016/j.ncl.2013.04.006
- Bird TD. Myotonic Dystrophy Type 1. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1165/. Accessed October 14, 2020.
- Dolzhenko E, Bennett MF, Richmond PA, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020;21(1):102. Published 2020 Apr 28. doi:10.1186/s13059-020-02017-z
- Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics. 2019;35(22):4754-4756. doi:10.1093/bioinformatics/btz431
- Ibanez K, Polke J, Hagelstrom T, et al. Whole genome sequencing for diagnosis of neurological repeat expansion disorders. bioRxiv 2020.11.06.371716; doi: https://doi.org/10.1101/2020.11.06.371716
- Roy S, Coldren C, Karunamurthy A, et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn. 2018;20(1):4-27. doi:10.1016/j.jmoldx.2017.11.003
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017;77(21):e31-e34. doi:10.1158/0008-5472.CAN-17-0337
- Dolzhenko E, Weisburd B, Ibanez Garikano K, et al. REViewer: Haplotype-resolved visualization of read alignments in and around tandem repeats. bioRxiv 2021.10.20.465046; doi: https://doi.org/10.1101/2021.10.20.465046