Introduction
Next-generation sequencing (NGS) technology has transformed the way researchers perform population genetics studies by making whole-genome sequencing (WGS) and complete variant information readily accessible. However, the generation of high-coverage sequencing data from hundreds to thousands of samples necessary for well-powered genome-wide association studies (GWAS) significantly drives up the cost, thus limiting its potential. Recently, low-coverage WGS followed by imputation has emerged as a cost-effective yet useful approach for capturing common and low-frequency variation,1-3 particularly in populations not well-represented in standard single nucleotide polymorphism (SNP) arrays.4
Genotype imputation refines data using information from reference haplotype panels to fill in gaps between sparsely mapped reads in data sets with low sequencing depth. This method enables researchers to infer untyped variants in a large sample of individuals, potentially boosting the statistical power of GWAS. A critical step in genomic prediction is the accuracy of imputation, which can have a significant impact on downstream analyses. Key factors influencing imputation accuracy include size and positional coverage of the reference panel, imputation method, minor allele frequency of the variants being imputed, and the accuracy of haplotype phasing.5
We combined the imputation accuracy of the ‘genotype likelihoods imputation and phasing method’ (GLIMPSE)6 with software acceleration on the Illumina DRAGEN™ Bio-IT platform to optimize the biallelic SNP variant calling performance when imputing low-coverage sequencing data (Figure 1). Integration of the accelerated GLIMPSE imputation tool into the DRAGEN platform reduces computation time by 40% and offers scalability with an end-to-end pipeline using a single command on multiple chromosomes. Additionally, our imputation workflow leverages the Illumina IRPv1 reference panel to further boost variant calling performance. In this article, we discuss how the DRAGEN implementation of GLIMPSE decreases the total number of false calls and present data to demonstrate how the imputation table and number of input samples can boost imputation accuracy.
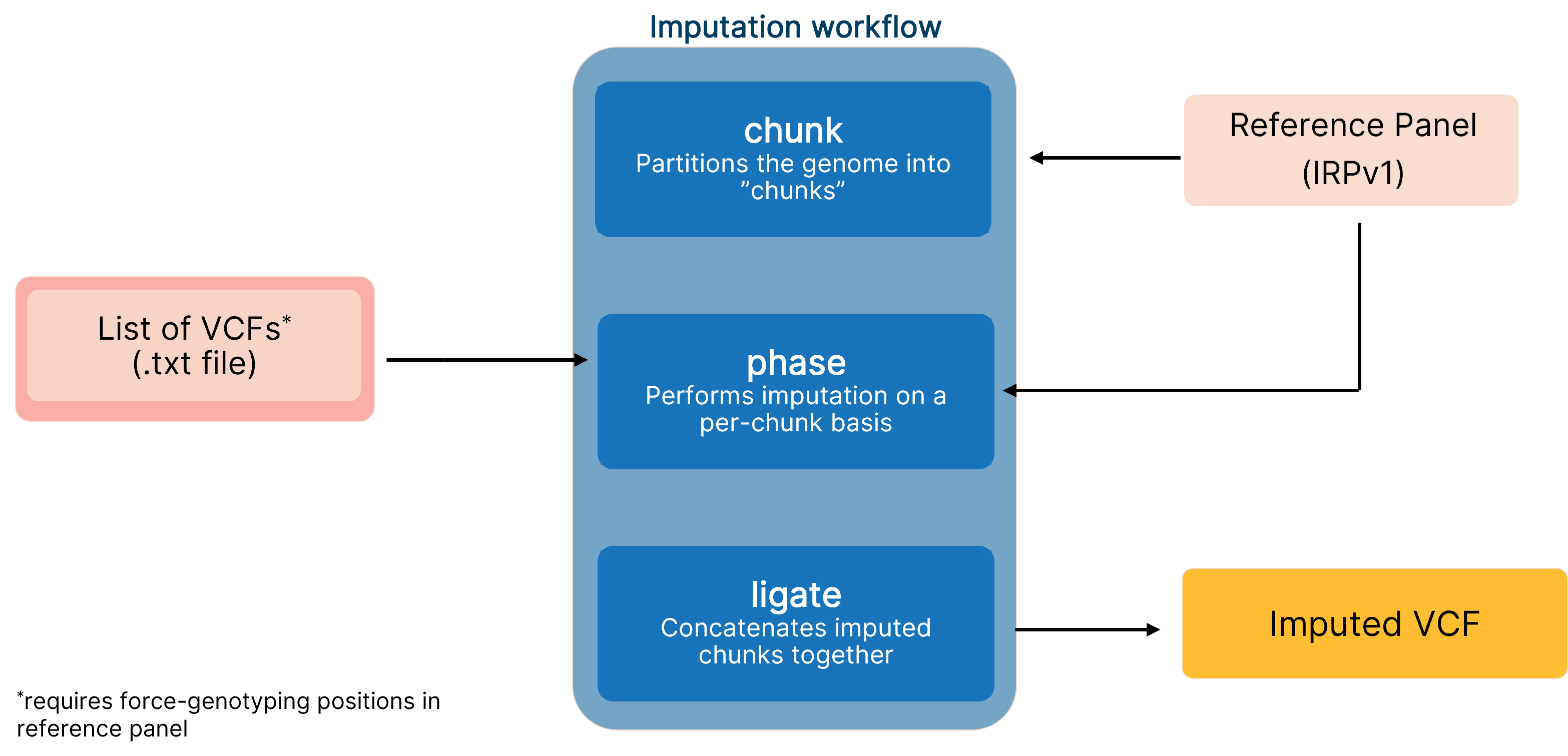
End-to-end pipeline for imputing low-coverage sequencing data using DRAGEN implementation of GLIMPSE, which leverages the IRPv1 reference panel for enhanced variant calling performance.
DRAGEN implementation of GLIMPSE improves variant calling performance in imputed 1× data
The imputation method or algorithm used directly influences the quality of imputed data.7 GLIMPSE is an open-source imputation software tool that produces high-quality consensus-based haplotype calling using a Gibbs sampler scheme at all variable positions in the reference panel.6 We deployed the highly accurate GLIMPSE on Illumina DRAGEN v4.0 to further accelerate the analysis timeline of this tool and improve imputation efficiency using a single command-line instruction. We demonstrate that the software accelerated code on DRAGEN v4.0 reduced imputation run time by 40% compared to GLIMPSE Phase v1.1.1 (Figure 2).
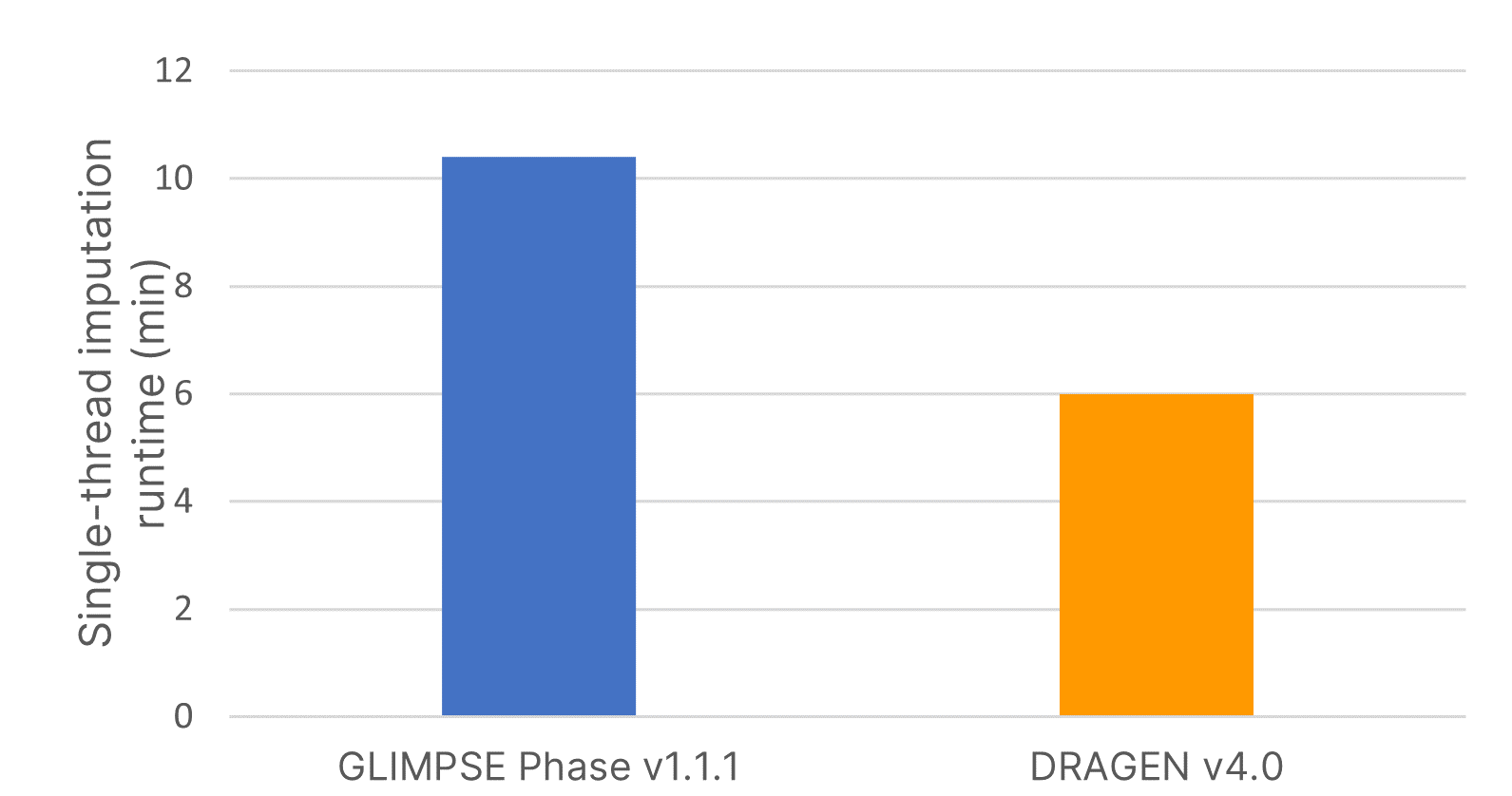
Imputation run times were assessed using input variant call format (VCF) files from 15 samples for imputing a region containing 83,172 positions using the large, diverse IRPv1 reference panel consisting of 2489 samples. Software-accelerated imputation reduced computational times by 1.7× compared to GLIMPSE Phase v1.1.1.
To assess general imputation performance, we used three samples HG002, HG003, and HG004 from the Genome-in-a-Bottle (GIAB) consortium,8,9 down-sampled to 1× coverage. These samples are well-characterized human genomes that have been widely used to validate sequencing pipelines and develop new variant calling methods. The truth data set consisted of NCBI benchmark SNPs (v4.2.1). 8,9
A comparison of the total false call rates between nonimputed and imputed low-coverage (1×) data from this small set of samples revealed a ~90% reduction in the false call rate in each individual sample imputed using the IRPv1 reference panel, largely due to a reduction in false negative (FN) calls (Figure 3). Our findings demonstrate that GLIMPSE deployed on DRAGEN v4.0 provides highly efficient genotype imputation and improved variant calling performance.
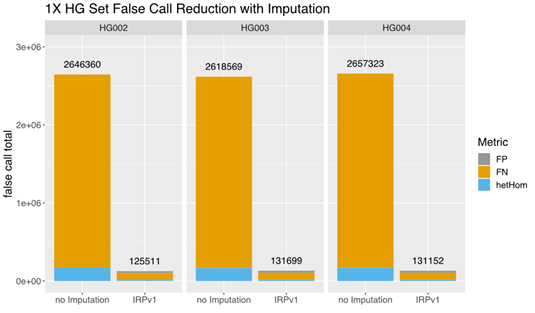
The total false call-rate was compared between low-coverage (1×) sequencing data with or without imputation using the GLIMPSE tool deployed on DRAGEN v4.0 with the IRPv1 reference panel. Imputation using this approach lowered the false call rate by ~90% in each sample (HG002, HG003, HG004) assessed. FP: false positive; FN: false negative; hetHom: heterozygous/homozygous ratio.
IRPv1 reference panel enhances imputation accuracy
Imputation is based on the principle of haplotype sharing to predict missing genotypes using a training set with high-confidence genotype information. As a result, the accuracy of any imputation tool is heavily dependent on the population structure of the reference panel used to infer untyped variants.7,10 We developed a new reference panel, IRPv1, and demonstrate the impact of the imputation reference panel size and genetic diversity on variant calling performance with GLIMPSE.
IRPv1 is an autosomal SNP reference panel containing 2489 samples from the 1000 Genomes Project, trimmed to exclude pediatric samples. These highly benchmarked samples have been variant called from the ~50× coverage New York Genome Center data against the GRCh38 human reference genome using DRAGEN v3.7.6. We excluded singleton SNPs and SNPs observed to be out of Hardy-Weinberg equilibrium. For SNPs where more than one alternative allele was observed, only the most frequently observed alternative allele was retained. SNPs were phased using SHAPEIT4.11
To assess the impact of reference panel attributes on variant calling performance, we tested three reference panels:
· IRPv1: large panel comprised of 2489 samples from 26 population groups
· IRPv1-100: small, high-density panel consisting of a subset of 100 samples from IRPv1
· IRPv1-PJL: genetically stratified small reference panel containing 96 samples from the Punjabi in Lahore, Pakistan (PJL) population
Five samples from three super populations—African (AFR), East Asian (EAS), and European (EUR)—were tested at 1× coverage with a truth dataset defined byof the same samples processed by DRAGEN at 50× coverage (Table 1). To ensure unbiased analyses, we excluded these 15 samples used for imputation from the IRPv1 reference panel. The subset of 100 samples in IRPv1-100 was selected such that the diversity across population groups in IPRv1 was maintained for this smaller panel.

AFR, African; EAS, East Asian; EUR, European.
The total number of positions represented in each panel are as follows:
· IRPv1: 49,493,544
· IRPv1-100: 22,066,395
· IRPv1-PJL: 13,429,942
Our results (Figure 4) demonstrate that the imputation performance was the highest with the complete IRPv1 reference panel, whereas the sensitivity was reduced with an increase in the false positive rate with the condensed panels. This reduction in performance was most pronounced in the AFR population, which contains more heterogeneity in the haplotype structure at the population level.7,12 Within this test population, the sensitivity was lowest, and the false positive rate was the highest when the IRPv1-PJL reference panel, which is genetically dissimilar from the AFR population, was used for imputation. These results underscore the importance of using a large and genetically diverse reference panel to maximize the likelihood that an observed haplotype is present in the reference data set, thus improving the overall imputation accuracy.
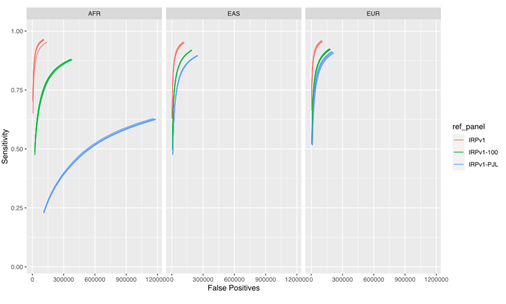
The sensitivity and false positive variant call rates were computed for three super populations (AFR, EAS, and EUR) using three reference panels (IRPv1, IRPv1-100, IRPv1-PJL). The overall imputation accuracy and variant calling performance was reliant on the size and genetic diversity of the reference panel used for inferring missing genotypes. AFR: African; EAS: East Asian; EUR: European populations.
Multisample mode boosts imputation performance
The GLIMPSE tool gives researchers the option to run their analyses in single or multisample modes to enhance variant calling performance using input data with low sequencing depth (1× coverage) even with a suboptimal reference panel. In multisample mode, a multisample VCF file is provided as input to the software. The imputation tool then imputes samples one at a time but incorporates other samples in the multisample VCF into the reference panel, using these samples as additional reference data points to aid in haplotype block definition and matching. We added an option for researchers to supply a list of single sample VCF files as input, which is then run in multisample mode as if a multisample VCF file had been provided, thus expanding the breadth of inputs that can be run in multisample mode.
To assess the impact of running samples individually or in multisample mode, we tested the five samples from the AFR super-population group described above using either the same three reference panels (IRPv1, IRPv1-100, IRPv1-PJL) or the genetically stratified IRPv1-PJL reference panel alone. The truth data set consisted of the same samples at 50× coverage. There was minimal impact of sample mode on imputation accuracy with larger, more diverse panels. However, our results demonstrate that applying multisample mode to low-coverage samples can boost sensitivity and improve imputation accuracy even with a less genetically diverse reference panel (Figure 5).
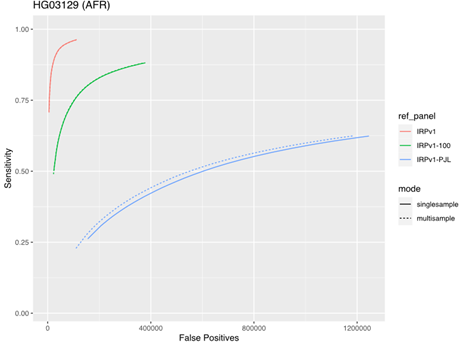
The sensitivity and false positive variant call rates were computed for five samples from the genetically diverse African (AFR) super population using three reference panels (IRPv1, IRPv1-100, IRPv1-PJL). Imputation was performed either in single- or multisample mode. The overall imputation accuracy and variant calling performance was boosted by running samples in multisample mode for the diverse AFR population even while using a suboptimal reference panel, though the best performance was obtained when running samples with a diverse panel.
Summary
In this article, we describe key enhancements to the GLIMPSE tool used for imputing genotypes from large low-coverage data sets. First, the highly accurate GLIMPSE software suite is embedded into DRAGEN v4.0 for accelerated analysis speeds and can be run using a single command-line instruction. Imputation using our modified workflow dramatically improves variant calling performance when imputing low coverage data. However, the overall imputation accuracy is reliant on an appropriate reference panel to infer unobserved genotypes. We developed a large, genetically diverse reference panel IRPv1, comprising 2489 samples from 26 sample groups, which when combined with our high-accuracy imputation tool, further enhanced variant calling performance tested in three super-populations. Using multisample mode further boosts imputation accuracy, particularly in if the reference panel is small and lacks sufficient diversity to capture all haplotypes present in the test population.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
References
1. Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11(7):499-511. doi:10.1038/nrg2796
2. Pasaniuc B, Rohland N, McLaren PJ, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44(6):631-635. doi:10.1038/ng.2283
3. Naj AC. Genotype Imputation in Genome-Wide Association Studies. Curr Protoc Hum Genet. 2019;102(1):e84. doi:10.1002/cphg.84
4. Martin AR, Atkinson EG, Chapman SB, et al. Low-coverage sequencing cost-effectively detects known and novel variation in underrepresented populations. Am J Hum Genet. 2021;108(4):656-668. doi:10.1016/j.ajhg.2021.03.012
5. Zhang B, Zhi D, Zhang K, Gao G, Limdi NN, Liu N. Practical Consideration of Genotype Imputation: Sample Size, Window Size, Reference Choice, and Untyped Rate. Stat Interface. 2011;4(3):339-352. doi: 10.4310/sii.2011.v4.n3.a8.
6. Rubinacci S, Ribeiro DM, Hofmeister RJ, Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat Genet. 2021;53(1):120-126. doi:10.1038/s41588-020-00756-0
7. Schurz H, Müller SJ, van Helden PD, et al. Evaluating the Accuracy of Imputation Methods in a Five-Way Admixed Population. Front Genet. 2019;10:34. doi:10.3389/fgene.2019.00034
8. Zook JM, Catoe D, McDaniel J, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data. 2016;3:160025. doi:10.1038/sdata.2016.25.
9. Wagner J, Olson ND, Harris L, et al. Benchmarking challenging small variants with linked and long reads. Cell Genomics. 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128.
10. Ahmad M, Sinha A, Ghosh S, et al. Inclusion of Population-specific Reference Panel from India to the 1000 Genomes Phase 3 Panel Improves Imputation Accuracy. Sci Rep. 2017;7(1):6733. doi:10.1038/s41598-017-06905-6
11. Delaneau O, Zagury JF, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10(1):5436. doi:10.1038/s41467-019-13225-y
12. Xu ZM, Rüeger S, Zwyer M, et al. Using population-specific add-on polymorphisms to improve genotype imputation in underrepresented populations. PLoS Comput Biol. 2022;18(1):e1009628. doi:10.1371/journal.pcbi.1009628