Microbial Genomics
Illumina next-generation sequencing takes you inside microbiology
Putting the big picture into focus by revealing the smallest of details with microbial genomics
Characterize unculturable microbes. Discover entirely new viruses. Develop new strategies to control outbreaks. Monitor host-pathogen interactions.
Next-generation sequencing (NGS) is opening new doors in microbial genomics, revealing fresh insight into how microbes impact humans and the environment.
- Learn how NGS enables a broad range of microbiology studies. Explore microbial sequencing methods.
- See how NGS is transforming infectious disease research, surveillance, characterization, and outbreak responses. Learn more about public health surveillance and infectious disease research.
- Learn about microbiome analysis for studying microbial communities. Or see how microbiome research provides scientists and pharmaceutical companies with data to refine drug discovery and development in this interview.
Through the power and high resolution of Illumina technology, you can now understand the genetic makeup of organisms that were previously impossible to study—helping you examine microbial biological functions, track genetic changes, rapidly respond to outbreaks, monitor food sources, and more.
Illumina offers technology and support to enhance your microbiology and infectious disease research. Learn more about NGS workflows and solutions based on your microbes of interest, sample types, and questions you want to answer with optimized end-to-end solutions. Read brochure (PDF).
Coronavirus sequencing
Next-generation sequencing is uniquely positioned in an infectious disease surveillance and outbreak model. Explore methods and find solutions to detect and characterize SARS-CoV-2 and other respiratory pathogens, track transmission routes, study co-infection, and investigate viral evolution.
Recent microbial genomics research

How genomics can help detect drug-resistant TB earlier
Around the world, next-generation sequencing has the power to address ongoing public health epidemics like tuberculosis
Read article
NGS is Revealing the Mysterious World of Microbes
Researchers are investigating the genomes of microbes to improve our understanding of human health, disease, and microbial evolution.
Read Interview
Tackling antimicrobial resistance, one genome at a time
Illumina’s pioneering sequencing tools are helping scientists combat this deadly threat across multiple continents
Read articleNGS Workflow Finder
Take the guesswork out of your next workflow. The NGS Workflow Finder provides personalized solution recommendations and resources so you can sequence with confidence.
Find your NGS workflow todayEmpowering access for groundbreaking genomic discoveries
Illumina benchtop sequencing systems are making NGS technology more accessible to laboratories worldwide. Learn how these systems provide the speed, power, and flexibility to make breakthroughs in microbiology, cancer research, and more. The MiSeq i100 Series or NextSeq 1000 and NextSeq 2000 Systems can help make your NGS research goals within reach.
Download eBook
Featured products

NovaSeq Reagent Kits
Reagent kits for the NovaSeq 6000 System provide ready-to-use cartridge-based reagents for cluster generation and SBS.
View Product
MiSeq i100 Series reagents
MiSeq i100 Series reagents combine the power of XLEAP-SBS chemistry with flexible, efficient, and sustainable workflows.
Learn More
Nextera XT and Nextera DNA Flex
Prepare sequencing libraries for small genomes, amplicons, plasmids, and other applications.
View ProductMiSeq i100 Series
Perform microbiology and infectious disease research using our fastest, simplest Illumina benchtop sequencer.
Learn More
In hot water with extremophiles
Dr Brian Hedlund uses genomics to study the microbes that populate hot springs. He is discovering new classes of organisms, learning how to culture microbes that would not have been culturable without genomic information, and learning new insights about microbial life.
View video
Educational resources
Beginner's guide to NGS
These educational resources cover key topics in next-generation sequencing designed for beginners. Learn the basics of NGS and find tips for getting started.
Illumina resources & tools
Find education and assistance for your genomics workflow, from start to finish. Find tools to glean insight from the data, then purchase what you need and get access to support.
Interested in receiving newsletters, case studies, and information on microbial genomics? Enter your email address.
Additional resources
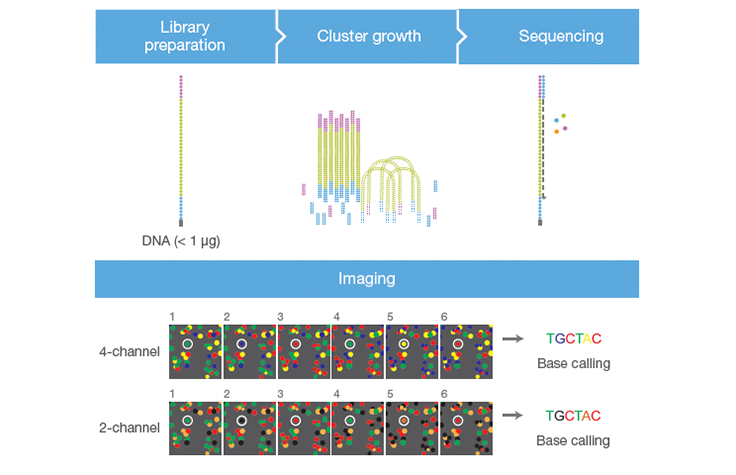
2-channel SBS technology
Learn how Illumina 2-channel sequencing by synthesis SBS technology delivers accurate, high-throughput sequencing with a simplified, cost-efficient design. This innovative chemistry powers a range of Illumina platforms, enabling reliable results for diverse genomic applications.

Introduction to the MiSeq i100 Series
Watch this webinar to get a comprehensive overview of the MiSeq i100 Series, which covers the overall system workflow, analytical applications, and performance.
Overview: NGS for Microbiology
View an introduction to NGS and its applications for microbiology.
Library Prep and Array Kit Selector
Find the right sequencing library prep kit or microarray for your needs. Compare similar kits or view further details.

Automated Methods for Metagenomic Analyses
Dr Russ Carmical discusses the benefits of using automated 16S rRNA and whole-genome sequencing methods for microbial genomics.
NGS for HIV Tropism Studies
Deep sequencing enables microbiology researchers to study HIV tropism and how it changes during infection.

TELL-Seq: Bringing Speed and Accuracy to Long-Range Sequencing
TELL-Seq technology is a simple library prep solution which enables Illumina NGS systems to generate highly accurate and economical long-range sequencing information.