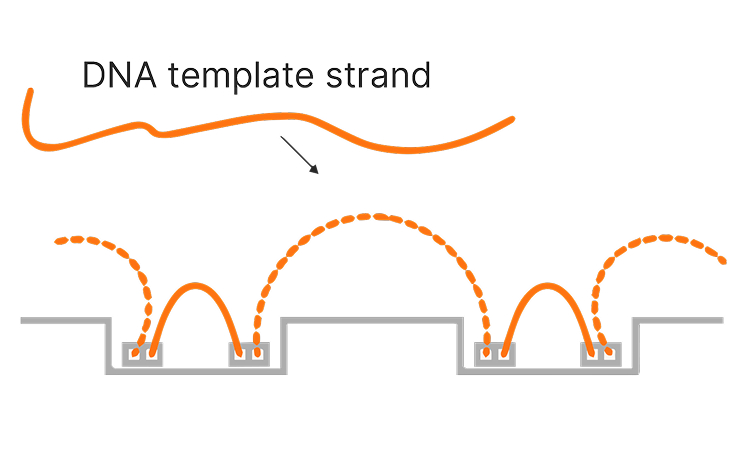
Illumina sequencing has significantly advanced genome mapping methods, helping researchers achieve highly accurate coverage over the vast majority of the human genome.1 However, for a small percentage of the genome, mapping short reads to a reference genome remains challenging. These challenges primarily occur in repetitive or low-complexity regions, regions with high sequence homology, or large structural variations.
Proximity mapped read technology enables improved mapping in difficult-to-map regions of the genome, ultralong phasing of genetic variants, and enhanced detection of structural variants.